Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Review Article
- Published: 06 June 2016

Determinants of genetic diversity
- Hans Ellegren 1 &
- Nicolas Galtier 2
Nature Reviews Genetics volume 17 , pages 422–433 ( 2016 ) Cite this article
29k Accesses
406 Citations
175 Altmetric
Metrics details
- Evolutionary biology
- Evolutionary genetics
- Genetic variation
- Molecular evolution
- Next-generation sequencing
This article has been updated
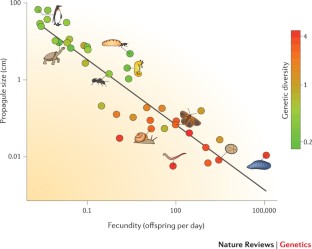
Lewontin's paradox — the much larger variation in species abundance than in genetic diversity — is closer to being explained.
The reproductive strategy of species has an impact on genome-wide diversity, providing a connection between population dynamic processes and the long-term effective population size ( N e ).
Selection at linked sites also affects genome-wide diversity, but not to an extent that it is sufficient alone to explain Lewontin's paradox.
Selection and demography, among other factors, contribute to variation in N e within genomes and leads to variation in diversity in different genomic regions of the same species.
Genetic polymorphism varies among species and within genomes, and has important implications for the evolution and conservation of species. The determinants of this variation have been poorly understood, but population genomic data from a wide range of organisms now make it possible to delineate the underlying evolutionary processes, notably how variation in the effective population size ( N e ) governs genetic diversity. Comparative population genomics is on its way to providing a solution to 'Lewontin's paradox' — the discrepancy between the many orders of magnitude of variation in population size and the much narrower distribution of diversity levels. It seems that linked selection plays an important part both in the overall genetic diversity of a species and in the variation in diversity within the genome. Genetic diversity also seems to be predictable from the life history of a species.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
176,64 € per year
only 14,72 € per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others

Opportunities and challenges of macrogenetic studies

Polygenic adaptation: a unifying framework to understand positive selection

Correlational selection in the age of genomics
Change history, 08 june 2016.
In the original version of this article, the author name in reference 73 (Stebbins, G. L. Self fertilization and population variability in the higher plants. Am. Naturalist 91 , 41–46 (1957)) was mis-spelled. This has now been corrected. The authors apologise for this error.
Lewontin, R. C. & Hubby, J. L. A molecular approach to the study of genic heterozygosity in natural populations. II. Amount of variation and degree of heterozygosity in natural populations of Drosophila pseudoobscura . Genetics 54 , 595–609 (1966).
PubMed PubMed Central CAS Google Scholar
Harris, H. Enzyme polymorphisms in man. Proc. R. Soc. Lond. B 164 , 298–310 (1966).
Article CAS PubMed Google Scholar
Quintana-Murci, L. & Clark, A. G. Population genetic tools for dissecting innate immunity in humans. Nat. Rev. Immunol. 13 , 280–293 (2013).
Article PubMed PubMed Central CAS Google Scholar
Bodmer, W. Genetic characterization of human populations: from ABO to a genetic map of the British people. Genetics 199 , 267–279 (2015).
Hake, S. & Ross-Ibarra, J. Genetic, evolutionary and plant breeding insights from the domestication of maize. eLife 4 , e05861 (2015).
Article PubMed Central CAS Google Scholar
Soares, M. P. & Weiss, G. The Iron Age of host–microbe interactions. EMBO Rep. 16 , 1482–1500 (2015).
Vander Wal, E., Garant, D., Festa-Bianchet, M. & Pelletier, F. Evolutionary rescue in vertebrates: evidence, applications and uncertainty. Phil. Trans. R. Soc. B 368 , 20120090 (2012).
Article Google Scholar
Forcada, J. & Hoffman, J. I. Climate change selects for heterozygosity in a declining fur seal population. Nature 511 , 462–465 (2014).
Begun, D. J. et al. Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans . PLoS Biol. 5 , e310 (2007).
Lack, J. B. et al. The Drosophila genome nexus: a population genomic resource of 623 Drosophila melanogaster genomes, including 197 from a single ancestral range population. Genetics 199 , 1229–1241 (2015).
McVean, G., Spencer, C. C. A. & Chaix, R. Perspectives on human genetic variation from the HapMap project. PLoS Genet. 1 , e54 (2005).
The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526 , 68–74 (2015).
Tenaillon, M. I. et al. Patterns of DNA sequence polymorphism along chromosome 1 of maize ( Zea mays ssp. mays L.). Proc. Natl Acad. Sci. USA 98 , 9161–9166 (2001).
Article CAS PubMed PubMed Central Google Scholar
Nordborg, M. et al. The pattern of polymorphism in Arabidopsis thaliana . PLoS Biol. 3 , e196 (2005).
Doniger, S. W. et al. A catalog of neutral and deleterious polymorphism in yeast. PLoS Genet. 4 , e1000183 (2008).
Wong, G. K. S. et al. A genetic variation map for chicken with 2.8 million single-nucleotide polymorphisms. Nature 432 , 717–722 (2004).
Sachidanandam, R. et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409 , 928–933 (2001).
Hodgkinson, A. & Eyre-Walker, A. Variation in the mutation rate across mammalian genomes. Nat. Rev. Genet. 12 , 756–766 (2011).
Lynch, M. Evolution of the mutation rate. Trends Genet. 26 , 345–352 (2010).
Charlesworth, B. Effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet. 10 , 195–205 (2009).
Kimura, M. The Neutral Theory of Molecular Evolution (Cambridge Univ. Press, 1983).
Book Google Scholar
Lewontin, R. The Genetic Basis of Evolutionary Change (Columbia Univ. Press, 1974). This book is a remarkably clear and early introduction to the problem of variation in genetic diversity and the first statement of the so-called Lewontin's paradox.
Google Scholar
Leffler, E. M. et al. Revisiting an old riddle: what determines genetic diversity levels within species? PLoS Biol. 10 , e1001388 (2012). This article contains a thorough review of the distribution of DNA sequence diversity across hundreds of eukaryotic species.
Reed, D. H. & Frankham, R. Correlation between fitness and genetic diversity. Conserv. Biol. 17 , 230–237 (2003).
Reed, D. H. & Frankham, R. How closely correlated are molecular and quantitative measures of genetic variation? A meta-analysis. Evolution 55 , 1095–1103 (2001).
Bjørnstad, O. N. & Grenfell, B. T. Noisy clockwork: time series analysis of population fluctuations in animals. Science 293 , 638–643 (2001).
Article PubMed Google Scholar
Sun, J., Cornelius, S. P., Janssen, J., Gray, K. A. & Motter, A. E. Regularity underlies erratic population abundances in marine ecosystems. J. R. Soc. Interface 12 , 20150235 (2015).
Article PubMed PubMed Central Google Scholar
Banks, S. C. et al. How does ecological disturbance influence genetic diversity? Trends Ecol. Evol. 28 , 670–679 (2013).
Alcala, N. & Vuilleumier, S. Turnover and accumulation of genetic diversity across large time-scale cycles of isolation and connection of populations. Proc. R. Soc. B 281 , 20141369 (2014).
Mayr, E. Animal Species and Evolution (Harvard Univ. Press, 1963).
Hewitt, G. The genetic legacy of the Quaternary ice ages. Nature 405 , 907–913 (2000).
Stuessy, T. F., Takayama, K., López-Sepúlveda, P. & Crawford, D. J. Interpretation of patterns of genetic variation in endemic plant species of oceanic islands. Bot. J. Linnean Soc. 174 , 276–288 (2014).
Aguilar, R., Quesada, M., Ashworth, L., Herrerias-Diego, Y. & Lobo, J. Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches. Mol. Ecol. 17 , 5177–5188 (2008).
Caplins, S. A. et al. Landscape structure and the genetic effects of a population collapse. Proc. R. Soc. B 281 , 20141798 (2014).
Coltman, D. W. Molecular ecological approaches to studying the evolutionary impact of selective harvesting in wildlife. Mol. Ecol. 17 , 221–235 (2008).
Lynch, M. The Origins of Genome Architecture (Sinauer Associates, 2007).
Romiguier, J. et al. Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature 515 , 261–263 (2014). This study shows a comparative analysis of patterns of diversity across animals revealing a strong influence of the life-history traits of species.
Sung, W., Ackerman, M. S., Miller, S. F., Doak, T. G. & Lynch, M. Drift-barrier hypothesis and mutation-rate evolution. Proc. Natl Acad. Sci. USA 109 , 18488–18492 (2012).
Ness, R. W., Morgan, A. D., Vasanthakrishnan, R. B., Colegrave, N. & Keightley, P. D. Extensive de novo mutation rate variation between individuals and across the genome of Chlamydomonas reinhardtii . Genome Res. 25 , 1739–1749 (2015).
Wright, S. Size of population and breeding structure in relation to evolution. Science 87 , 430–431 (1938).
Weber, D., Stewart, B. S., Garza, J. C. & Lehman, N. An empirical genetic assessment of the severity of the northern elephant seal population bottleneck. Curr. Biol. 10 , 1287–1290 (2000).
Hedrick, P. W. Conservation genetics and North American bison ( Bison bison ). J. Hered. 100 , 411–420 (2009).
Spielman, D., Brook, B. W. & Frankham, R. Most species are not driven to extinction before genetic factors impact them. Proc. Natl Acad. Sci. USA 101 , 15261–15264 (2004).
Nabholz, B., Mauffrey, J. -F., Bazin, E., Galtier, N. & Glemin, S. Determination of mitochondrial genetic diversity in mammals. Genetics 178 , 351–361 (2008).
McCusker, M. R. & Bentzen, P. Positive relationships between genetic diversity and abundance in fishes. Mol. Ecol. 19 , 4852–4862 (2010).
Perry, G. H. et al. Comparative RNA sequencing reveals substantial genetic variation in endangered primates. Genome Res. 22 , 602–610 (2012).
Pinsky, M. L. & Palumbi, S. R. Meta-analysis reveals lower genetic diversity in overfished populations. Mol. Ecol. 23 , 29–39 (2014).
Ho, S. Y. W. & Shapiro, B. Skyline-plot methods for estimating demographic history from nucleotide sequences. Mol. Ecol. Resour. 11 , 423–434 (2011).
Drummond, A. J., Rambaut, A., Shapiro, B. & Pybus, O. G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22 , 1185–1192 (2005).
Li, H. & Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 475 , 493–496 (2011).
Liu, X. & Fu, Y. -X. Exploring population size changes using SNP frequency spectra. Nat. Genet. 47 , 555–559 (2015).
Schiffels, S. & Durbin, R. Inferring human population size and separation history from multiple genome sequences. Nat. Genet. 46 , 919–925 (2014).
Nadachowska-Brzyska, K., Li, C., Smeds, L., Zhang, G. & Ellegren, H. Temporal dynamics of avian populations during Pleistocene revealed by whole-genome sequences. Curr. Biol. 25 , 1375–1380 (2015).
Jarne, P. Mating system, bottlenecks and genetic polymorphism in hermaphroditic animals. Genet. Res. 65 , 193–207 (1995).
Charlesworth, D. & Wright, S. Breeding systems and genome evolution. Curr. Opin. Genet. Dev. 11 , 685–690 (2001).
Glémin, S., Bazin, E. & Charlesworth, D. Impact of mating systems on patterns of sequence polymorphism in flowering plants. Proc. R. Soc. B 273 , 3011–3019 (2006).
Glémin, S. & Muyle, A. Mating systems and selection efficacy: a test using chloroplastic sequence data in angiosperms. J. Evol. Biol. 27 , 1386–1399 (2014).
Hartfield, M. Evolutionary genetic consequences of facultative sex and outcrossing. J. Evol. Biol. 29 , 5–22 (2016). This review discusses the theoretical predictions and empirical evidence regarding genome evolution in asexual versus sexual contexts.
Slotte, T. et al. The Capsella rubella genome and the genomic consequences of rapid mating system evolution. Nat. Genet. 45 , 831–835 (2013).
Burgarella, C. et al. Molecular evolution of freshwater snails with contrasting mating systems. Mol. Biol. Evol. 32 , 2403–2416 (2015).
Thomas, C. G. et al. Full-genome evolutionary histories of selfing, splitting, and selection in Caenorhabditis . Genome Res. 25 , 667–678 (2015).
Dey, A., Chan, C. K. W., Thomas, C. G. & Cutter, A. D. Molecular hyperdiversity defines populations of the nematode Caenorhabditis brenneri . Proc. Natl Acad. Sci. USA 110 , 11056–11060 (2013).
Dolgin, E. S., Charlesworth, B. & Cutter, A. D. Population frequencies of transposable elements in selfing and outcrossing Caenorhabditis nematodes. Genet. Res. 90 , 317–329 (2008).
Article CAS Google Scholar
Wright, S. I., Kalisz, S. & Slotte, T. Evolutionary consequences of self-fertilization in plants. Proc. R. Soc. B 280 , 20130133 (2013).
Balloux, F., Lehmann, L. & de MeeÛs, T. The population genetics of clonal and partially clonal diploids. Genetics 164 , 1635–1644 (2003).
PubMed PubMed Central Google Scholar
Mark Welch, D. B. & Meselson, M. Evidence for the evolution of Bdelloid rotifers without sexual reproduction or genetic exchange. Science 288 , 1211–1215 (2000).
Delmotte, F. et al. Phylogenetic evidence for hybrid origins of asexual lineages in an aphid species. Evolution 57 , 1291–1303 (2003).
Schaefer, I. et al. No evidence for the 'Meselson effect' in parthenogenetic oribatid mites (Oribatida, Acari). J. Evol. Biol. 19 , 184–193 (2006).
Schwander, T., Henry, L. & Crespi Bernard, J. Molecular evidence for ancient asexuality in Timema stick insects. Curr. Biol. 21 , 1129–1134 (2011).
Hollister, J. D. et al. Recurrent loss of sex is associated with accumulation of deleterious mutations in Oenothera . Mol. Biol. Evol. 32 , 896–905 (2015).
Maynard Smith, J. The Evolution of Sex (Cambridge Univ. Press, 1978).
McDonald, M. J., Rice, D. P. & Desai, M. M. Sex speeds adaptation by altering the dynamics of molecular evolution. Nature 531 , 233–236 (2016).
Stebbins, G. L. Self fertilization and population variability in the higher plants. Am. Naturalist 91 , 41–46 (1957).
Judson, O. P. & Normark, B. B. Ancient asexual scandals. Trends Ecol. Evol. 11 , 41–46 (1996).
Simon, J. C., Delmotte, F., Rispe, C. & Crease, T. Phylogenetic evidence for hybrid origins of asexual lineages in an aphid species. Evolution 57 , 1291–1303 (2003).
Igic, B. & Busch, J. W. Is self-fertilization an evolutionary dead end? New Phytol. 198 , 386–397 (2013).
Tajima, F. Relationship between DNA polymorphism and fixation time. Genetics 125 , 447–454 (1990).
Cutter, A. D. & Payseur, B. A. Genomic signatures of selection at linked sites: unifying the disparity among species. Nat. Rev. Genet. 14 , 262–274 (2013).
Maynard Smith, J. & Haigh, J. The hitch-hiking effect of a favourable gene. Genet. Res. 23 , 23–35 (1974).
Kaplan, N. L., Hudson, R. R. & Langley, C. H. The “hitchhiking effect” revisited. Genetics 123 , 887–899 (1989).
Gillespie, J. H. Genetic drift in an infinite population: the pseudohitchhiking model. Genetics 155 , 909–919 (2000).
Gillespie, J. H. Is the population size of a species relevant to its evolution? Evolution 55 , 2161–2169 (2001). This paper shows a theoretical examination of the effects of recurrent adaptive substitutions on linked loci and their relationship to N e .
Charlesworth, B., Morgan, M. T. & Charlesworth, D. The effect of deleterious mutations on neutral molecular variation. Genetics 134 , 1289–1303 (1993). This study shows a theoretical examination of the effects of recurrent deleterious substitutions on linked loci and the background selection model.
Charlesworth, B. The effect of background selection against deleterious mutations on weakly selected, linked variants. Genet. Res. 63 , 213–227 (1994).
Corbett-Detig, R. B., Hartl, D. L. & Sackton, T. B. Natural selection constrains neutral diversity across a wide range of species. PLoS Biol. 13 , e1002112 (2015). This article demonstrates the role of linked selection in shaping the within-genome variation in polymorphism and its relationship with N e .
Coop, G. Does linked selection explain the narrow range of genetic diversity across species? bioRxiv http://dx.doi.org/10.1101/042598 (2016).
Elyashiv, E. et al. A genomic map of the effects of linked selection in Drosophila . arXiv http://arXiv.org//abs/1408.5461v1 (2014).
Comeron, J. M. Background selection as baseline for nucleotide variation across the Drosophila genome. PLoS Genet. 10 , e1004434 (2014).
Enard, D., Messer, P. W. & Petrov, D. A. Genome-wide signals of positive selection in human evolution. Genome Res. 24 , 885–895 (2014).
Gossmann, T. I., Woolfit, M. & Eyre-Walker, A. Quantifying the variation in the effective population size within a genome. Genetics 189 , 1389–1402 (2011).
Wu, C.-I. The genic view of the process of speciation. J. Evol. Biol. 14 , 851–865 (2001).
Begun, D. J. & Aquadro, C. F. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster . Nature 356 , 519–520 (1992).
Nachman, M. W. Single nucleotide polymorphisms and recombination rate in humans. Trends Genet. 17 , 481–485 (2001).
Lercher, M. J. & Hurst, L. D. Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet. 18 , 337–340 (2002).
Dvorak, J., Luo, M. C. & Yang, Z. L. Restriction fragment length polymorphism and divergence in the genomic regions of high and low recombination in self-fertilizing and cross-fertilizing Aegilops species. Genetics 148 , 423–434 (1998).
Stephan, W. & Langley, C. H. DNA polymorphism in Lycopersicon and crossing-over per physical length. Genetics 150 , 1585–1593 (1998).
Cutter, A. D. & Choi, J. Y. Natural selection shapes nucleotide polymorphism across the genome of the nematode Caenorhabditis briggsae . Genome Res. 20 , 1103–1111 (2010).
Fay, J. C. & Wu, C. I. Hitchhiking under positive Darwinian selection. Genetics 155 , 1405–1413 (2000).
Campos, J. L., Halligan, D. L., Haddrill, P. R. & Charlesworth, B. The relation between recombination rate and patterns of molecular evolution and variation in Drosophila melanogaster . Mol. Biol. Evol. 31 , 1010–1028 (2014).
Messer, P. W. & Petrov, D. A. Frequent adaptation and the McDonald–Kreitman test. Proc. Natl Acad. Sci. USA 110 , 8615–8620 (2013).
Sella, G., Petrov, D. A., Przeworski, M. & Andolfatto, P. Pervasive natural selection in the Drosophila genome? PLoS Genet. 5 , e1000495 (2009). This article reviews the evidence for a pervasive role of linked selection on patterns of genetic variation in Drosophila species.
Slotte, T. The impact of linked selection on plant genomic variation. Brief. Funct. Genomics 13 , 268–275 (2014).
Lohmueller, K. E. et al. Natural selection affects multiple aspects of genetic variation at putatively neutral sites across the human genome. PLoS Genet. 7 , e1002326 (2011).
Messer, P. W. SLiM: simulating evolution with selection and linkage. Genetics 194 , 1037–1039 (2013).
Hernandez, R. D. A flexible forward simulator for populations subject to selection and demography. Bioinformatics 24 , 2786–2787 (2008).
Bank, C., Ewing, G. B., Ferrer-Admettla, A., Foll, M. & Jensen, J. D. Thinking too positive? Revisiting current methods of population genetic selection inference. Trends Genet. 30 , 540–546 (2014).
Coop, G. & Ralph, P. Patterns of neutral diversity under general models of selective sweeps. Genetics 192 , 205–224 (2012).
Bolívar, P., Mugal, C. F., Nater, A. & Ellegren, H. Recombination rate variation modulates gene sequence evolution mainly via GC-biased gene conversion, not Hill–Robertson interference, in an avian system. Mol. Biol. Evol. 33 , 216–227 (2016).
Payseur, B. A. & Nachman, M. W. Gene density and human nucleotide polymorphism. Mol. Biol. Evol. 19 , 336–340 (2002).
Charlesworth, B. Background selection and patterns of genetic diversity in Drosophila melanogaster . Genet. Res. 68 , 131–149 (1996).
Hudson, R. R. & Kaplan, N. L. Deleterious background selection with recombination. Genetics 141 , 1605–1617 (1995).
Nordborg, M., Charlesworth, B. & Charlesworth, D. The effect of recombination on background selection. Genet. Res. 67 , 159–174 (1996).
Flowers, J. M. et al. Natural selection in gene-dense regions shapes the genomic pattern of polymorphism in wild and domesticated rice. Mol. Biol. Evol. 29 , 675–687 (2012).
Burri, R. et al. Linked selection and recombination rate variation drive the evolution of the genomic landscape of differentiation across the speciation continuum of Ficedula flycatchers. Genome Res. 25 , 1656–1665 (2015). This study is a high-resolution examination of genome-wide patterns of diversity and the role of recombination and linked selection in several species of flycatcher.
Nabholz, B. et al. Transcriptome population genomics reveals severe bottleneck and domestication cost in the African rice ( Oryza glaberrima ). Mol. Ecol. 23 , 2210–2227 (2014).
Hellmann, I., Ebersberger, I., Ptak, S. E., Pääbo, S. & Przeworski, M. A neutral explanation for the correlation of diversity with recombination rates in humans. Am. J. Hum. Genet. 72 , 1527–1535 (2003).
Yang, S. et al. Parent-progeny sequencing indicates higher mutation rates in heterozygotes. Nature 523 , 463–467 (2015).
Arbeithuber, B., Betancourt, A. J., Ebner, T. & Tiemann-Boege, I. Crossovers are associated with mutation and biased gene conversion at recombination hotspots. Proc. Natl Acad. Sci. USA 112 , 2109–2114 (2015).
Rattray, A., Santoyo, G., Shafer, B. & Strathern, J. N. Elevated mutation rate during meiosis in Saccharomyces cerevisiae . PLoS Genet. 11 , e1004910 (2015).
Duret, L. & Galtier, N. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu. Rev. Genom. Hum. Genet. 10 , 285–311 (2009).
Wallberg, A., Glémin, S. & Webster, M. T. Extreme recombination frequencies shape genome variation and evolution in the honeybee, Apis mellifera . PLoS Genet. 11 , e1005189 (2015).
Hammer, M. F. et al. The ratio of human X chromosome to autosome diversity is positively correlated with genetic distance from genes. Nat. Genet. 42 , 830–831 (2010).
Arbiza, L., Gottipati, S., Siepel, A. & Keinan, A. Contrasting X-linked and autosomal diversity across 14 human populations. Am. J. Hum. Genet. 94 , 827–844 (2014).
Gottipati, S., Arbiza, L., Siepel, A., Clark, A. G. & Keinan, A. Analyses of X-linked and autosomal genetic variation in population-scale whole genome sequencing. Nat. Genet. 43 , 741–743 (2011).
Charlesworth, B. The role of background selection in shaping patterns of molecular evolution and variation: evidence from variability on the Drosophila X chromosome. Genetics 191 , 233–246 (2012).
Frankham, R. How closely does genetic diversity in finite populations conform to predictions of neutral theory? Large deficits in regions of low recombination. Heredity 108 , 167–178 (2012). This paper reviews and demonstrates the reduction in genetic diversity in low-recombining genomic regions, including sex chromosomes, in plants and animals.
Mank, J. E., Vicoso, B., Berlin, S. & Charlesworth, B. Effective population size and the faster-X effect: empirical results and their interpretation. Evolution 64 , 663–674 (2010).
Corl, A. & Ellegren, H. The genomic signature of sexual selection in the genetic diversity of the sex chromosomes and autosomes. Evolution 66 , 2138–2149 (2012).
Huang, H. & Rabosky, D. L. Sex-linked genomic variation and its relationship to avian plumage dichromatism and sexual selection. BMC Evol. Biol. 15 , 199 (2015).
Smeds, L. et al. Genomic identification and characterization of the pseudoautosomal region in highly differentiated avian sex chromosomes. Nat. Commun. 5 , 5448 (2014).
Article PubMed CAS Google Scholar
Lien, S., Szyda, J., Schechinger, B., Rappold, G. & Arnheim, N. Evidence for heterogeneity in recombination in the human pseudoautosomal region: high resolution analysis by sperm typing and radiation-hybrid mapping. Am. J. Hum. Genet. 66 , 557–566 (2000).
Bussell, J. J., Pearson, N. M., Kanda, R., Filatov, D. A. & Lahn, B. T. Human polymorphism and human–chimpanzee divergence in pseudoautosomal region correlate with local recombination rate. Gene 368 , 94–100 (2006).
Charlesworth, B. & Charlesworth, D. The degeneration of Y chromosomes. Phil. Trans. R. Soc. Lond. B 355 , 1563–1572 (2000).
Bachtrog, D. Y-chromosome evolution: emerging insights into processes of Y-chromosome degeneration. Nat. Rev. Genet. 14 , 113–124 (2013).
Mank, J. E. Small but mighty: the evolutionary dynamics of W and Y sex chromosomes. Chromosome Res. 20 , 21–33 (2011).
Hellborg, L. & Ellegren, H. Low levels of nucleotide diversity in mammalian Y chromosomes. Mol. Biol. Evol. 21 , 158–163 (2004).
Bachtrog, D., Thornton, K., Clark, A., Andolfatto, P. & Harrison, R. Extensive introgression of mitochondrial DNA relative to nuclear genes in the Drosophila yakuba species group. Evolution 60 , 292–302 (2006).
Shen, P. et al. Population genetic implications from sequence variation in four Y chromosome genes. Proc. Natl Acad. Sci. USA 97 , 7354–7359 (2000).
Qiu, S., Bergero, R., Forrest, A., Kaiser, V. B. & Charlesworth, D. Nucleotide diversity in Silene latifolia autosomal and sex-linked genes. Proc. R. Soc. B 277 , 3283–3290 (2010).
Filatov, D. A., Laporte, V., Vitte, C. & Charlesworth, D. DNA diversity in sex-linked and autosomal genes of the plant species Silene latifolia and Silene dioica . Mol. Biol. Evol. 18 , 1442–1454 (2001).
Smeds, L. et al. Evolutionary analysis of the female-specific avian W chromosome. Nat. Commun. 6 , 7330 (2015).
Wilson Sayres, M. A., Lohmueller, K. E. & Nielsen, R. Natural selection reduced diversity on human Y chromosomes. PLoS Genet. 10 , e1004064 (2014).
Ellegren, H. Characteristics, causes and evolutionary consequences of male-biased mutation. Proc. R. Soc. B 274 , 1–10 (2007).
Kong, A. et al. Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467 , 1099–1103 (2010).
Venn, O. et al. Strong male bias drives germline mutation in chimpanzees. Science 344 , 1272–1275 (2014).
Cutter, A. D., Jovelin, R. & Dey, A. Molecular hyperdiversity and evolution in very large populations. Mol. Ecol. 22 , 2074–2095 (2013). This article discusses the specificities and challenges associated with very highly polymorphic species, with a focus on Caenorhabditis nematodes.
Drouin, G. Characterization of the gene conversions between the multigene family members of the yeast genome. J. Mol. Evol. 55 , 14–23 (2002).
Borts, R. H. & Haber, J. E. Meiotic recombination in yeast: alteration by multiple heterozygosities. Science 237 , 1459–1465 (1987).
Dobzhansky, T. Evolution, Genetics, and Man (Wiley, 1955).
Ohta, T. Slightly deleterious mutant substitutions in evolution. Nature 246 , 96–98 (1973).
Hubby, J. L. & Lewontin, R. C. A molecular approach to the study of genic heterozygosity in natural populations. I. The number of alleles at different loci in Drosophila pseudoobscura . Genetics 54 , 577–594 (1966).
Soulé, M. in Molecular Evolution (ed. Ayala, F.) 60–77 (Sinauer Associates, 1976).
Nevo, E., Beiles, A. & Ben-Shlomo, R. in Evolutionary Dynamics of Genetic Diversity: Proceedings of a Symposium held in Manchester, England, March 29–30, 1983 (ed. Mani, G. S.) (Springer, 1984).
Hamrick, J. L. & Godt, M. J. W. Effects of life history traits on genetic diversity in plant species. Phil. Trans. R. Soc. Lond. B 351 , 1291–1298 (1996).
Cole, C. T. Genetic variation in rare and common plants. Annu. Rev. Ecol. Evol. Systemat. 34 , 213–237 (2003).
Avise, J. C. et al. Intraspecific phylogeography: the mitochondrial DNA bridge between population genetics and systematics. Annu. Rev. Ecol. Systemat. 18 , 489–522 (1987).
Bazin, E., Glémin, S. & Galtier, N. Population size does not influence mitochondrial genetic diversity in animals. Science 312 , 570–572 (2006).
Nabholz, B., Glémin, S. & Galtier, N. The erratic mitochondrial clock: variations of mutation rate, not population size, affect mtDNA diversity across birds and mammals. BMC Evol. Biol. 9 , 1–13 (2009).
Ballard, J. W. O. & Whitlock, M. C. The incomplete natural history of mitochondria. Mol. Ecol. 13 , 729–744 (2004).
Berlin, S., Tomaras, D. & Charlesworth, B. Low mitochondrial variability in birds may indicate Hill–Robertson effects on the W chromosome. Heredity 99 , 389–396 (2007).
Hurst, G. D. D. & Jiggins, F. M. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc. R. Soc. B 272 , 1525–1534 (2005).
Galtier, N., Nabholz, B., Glémin, S. & Hurst, G. D. D. Mitochondrial DNA as a marker of molecular diversity: a reappraisal. Mol. Ecol. 18 , 4541–4550 (2009).
Piganeau, G. & Eyre-Walker, A. Evidence for variation in the effective population size of animal mitochondrial DNA. PLoS ONE 4 , e4396 (2009).
Jarne, P. & Lagoda, P. J. L. Microsatellites, from molecules to populations and back. Trends Ecol. Evol. 11 , 424–429 (1996).
Väli, Ü., Einarsson, A., Waits, L. & Ellegren, H. To what extent do microsatellite markers reflect genome-wide genetic diversity in natural populations? Mol. Ecol. 17 , 3808–3817 (2008).
Fungtammasan, A. et al. Accurate typing of short tandem repeats from genome-wide sequencing data and its applications. Genome Res. 25 , 736–749 (2015).
Ellegren, H. Genome sequencing and population genomics in non-model organisms. Trends Ecol. Evol. 29 , 51–63 (2014).
Lynch, M. & Conery, J. S. The origins of genome complexity. Science 302 , 1401–1404 (2003).
Wright, S. Evolution in Mendelian populations. Genetics 16 , 97–159 (1931).
Luikart, G., Ryman, N., Tallmon, D., Schwartz, M. & Allendorf, F. Estimation of census and effective population sizes: the increasing usefulness of DNA-based approaches. Conserv. Genet. 11 , 355–373 (2010).
Palstra, F. P. & Fraser, D. J. Effective/census population size ratio estimation: a compendium and appraisal. Ecol. Evol. 2 , 2357–2365 (2012).
Gilbert, K. J. & Whitlock, M. C. Evaluating methods for estimating local effective population size with and without migration. Evolution 69 , 2154–2166 (2015).
Browning, S. R. & Browning, B. L. Accurate non-parametric estimation of recent effective population size from segments of identity by descent. Am. J. Hum. Genet. 97 , 404–418 (2015).
Kirin, M. et al. Genomic runs of homozygosity record population history and consanguinity. PLoS ONE 5 , e13996 (2010).
Palamara, P. F., Lencz, T., Darvasi, A. & Pe'er, I. Length distributions of identity by descent reveal fine-scale demographic history. Am. J. Hum. Genet. 91 , 809–822 (2012).
Download references
Acknowledgements
This work was supported by Swedish Research Council grants (2010–5650 and 2013–8271), a European Research Council grant (AdG 249976) and the Knut and Alice Wallenberg Foundation to H.E., and by a European Research Council grant (AdG 232971) and a French National Research Agency grant (ANR-10-BINF-01-01) to N.G. The authors thank N. Bierne, S. Glemin and M. Lascoux for comments on the manuscript.
Author information
Authors and affiliations.
Department of Evolutionary Biology, Evolutionary Biology Centre, Uppsala University, Norbyvägen 18D, Uppsala, SE-753 36, Sweden
Hans Ellegren
Institute of Evolutionary Sciences, French National Centre for Scientific Research (CNRS), University of Montpellier 2, Place E. Bataillon, Montpellier, 34095, France
Nicolas Galtier
You can also search for this author in PubMed Google Scholar
Corresponding authors
Correspondence to Hans Ellegren or Nicolas Galtier .
Ethics declarations
Competing interests.
The authors declare no competing financial interests.
PowerPoint slides
Powerpoint slide for fig. 1, powerpoint slide for fig. 2, powerpoint slide for fig. 3, powerpoint slide for fig. 4.
(Also known as genetic polymorphism). Variation in a DNA sequence between distinct individuals (or chromosomes) of a given species (or population).
Allelic variants of proteins that can be separated by electrophoresis based on differences in charge or structure.
The complete spread of a mutation in the population such that it replaces all other alleles at a site.
Fluctuation of allele frequency among generations in a population owing to the randomness of survival and reproduction of individuals, irrespective of selective pressures.
( N e ). The number of breeding individuals in an idealized population that would show the same amount of genetic drift (or inbreeding, or any other variable of interest) as the population under consideration.
( N c ).The number of individuals in a population.
A form of selection in which the selective advantage or disadvantage of a genotype is dependent on its frequency relative to other genotypes.
A sharp and rapid reduction in the size of a population.
The probability that two randomly sampled gene copies in a population carry distinct alleles; a measure of the genetic diversity.
The idea, based on the concept of diminishing returns, that selection can only improve a trait up to a point at which the next incremental improvement will be overwhelmed by the power of genetic drift.
A retrospective model of the distribution of gene divergence in a genealogy.
Chromosomal segments carried by two or more individuals that are identical because they have been inherited from a common ancestor, without recombination.
A form of genome evolution in which the number of sets of chromosomes increases.
The non-random association of alleles at two loci, often but not always due to physical linkage on the same chromosome.
Elimination or reduction of genetic diversity in the neighbourhood of a beneficial allele that increases in frequency in the population, typically after an environmental change.
Selective sweeps in which the beneficial allele corresponds to a single, new mutation appearing after an environmental change.
Selective sweeps in which the beneficial allele exists before an environmental change (thus representing standing variation) and is initially neutral or even slightly deleterious, or appears several times independently.
Pervasive reduction of genetic diversity owing to recurrent selective sweeps.
Reduction of genetic diversity owing to selection against deleterious mutations at linked loci.
New alleles entering the population by hybridization with members of a differentiated population or even a different species.
The change in allele frequency at a locus that itself is not necessarily affected by selection but is genetically linked to a locus that is.
The distribution of the frequency of variants across biallelic loci in a population sample.
A mating system in which males mate with more than one female.
A mating system in which females mate with more than one male.
When an organism of a particular sex carries two different types of sex chromosomes: that is, males of many animals and plants and females of birds, some fish and lizards, butterflies, and others.
The situation when there is only one chromosome copy in an individual of a diploid species, as for the X chromosome in males of many species.
Rights and permissions
Reprints and permissions
About this article
Cite this article.
Ellegren, H., Galtier, N. Determinants of genetic diversity. Nat Rev Genet 17 , 422–433 (2016). https://doi.org/10.1038/nrg.2016.58
Download citation
Published : 06 June 2016
Issue Date : July 2016
DOI : https://doi.org/10.1038/nrg.2016.58
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by
Population status and genetic assessment of mugger (crocodylus palustris) in a tropical regulated river system in north india.
- Surya Prasad Sharma
- Mirza Ghazanfarullah Ghazi
- Syed Ainul Hussain
Scientific Reports (2024)
Population genetics of the endangered Clanwilliam sandfish Labeo seeberi: considerations for conservation management
- Clint Rhode
- Shaun F. Lesch
- Martine S. Jordaan
Aquatic Sciences (2024)
Microsatellite and mtDNA-based exploration of inter-generic hybridization and patterns of genetic diversity in major carps of Punjab, Pakistan
- Shakeela Parveen
- Khalid Abbas
- Laiba Shafique
Aquaculture International (2024)
Employing plant DNA barcodes for pomegranate species identification in Al-Baha Region, Saudi Arabia
- Fatima Omari Alzahrani
- Houda Maaroufi Dguimi
- Sonia Zaoui
Journal of Umm Al-Qura University for Applied Sciences (2024)
Metabolomic profiling of wild rooibos (Aspalathus linearis) ecotypes and their antioxidant-derived phytopharmaceutical potential
- C. Wilkinson
- N. P. Makunga
Metabolomics (2024)
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing newsletter — what matters in science, free to your inbox daily.

ORIGINAL RESEARCH article
Management of genetic diversity in the era of genomics.

- 1 Department of Animal and Aquacultural Sciences, Norwegian University of Life Sciences, Ås, Norway
- 2 NOFIMA, Ås, Norway
- 3 The Roslin Institute and R(D)SVS, The University of Edinburgh, Edinburgh, United Kingdom
Management of genetic diversity aims to (i) maintain heterozygosity, which ameliorates inbreeding depression and loss of genetic variation at loci that may become of importance in the future; and (ii) avoid genetic drift, which prevents deleterious recessives (e.g., rare disease alleles) from drifting to high frequency, and prevents random drift of (functional) traits. In the genomics era, genomics data allow for many alternative measures of inbreeding and genomic relationships. Genomic relationships/inbreeding can be classified into (i) homozygosity/heterozygosity based (e.g., molecular kinship matrix); (ii) genetic drift-based, i.e., changes of allele frequencies; or (iii) IBD-based, i.e., SNPs are used in linkage analyses to identify IBD segments. Here, alternative measures of inbreeding/relationship were used to manage genetic diversity in genomic optimal contribution (GOC) selection schemes. Contrary to classic inbreeding theory, it was found that drift and homozygosity-based inbreeding could differ substantially in GOC schemes unless diversity management was based upon IBD. When using a homozygosity-based measure of relationship, the inbreeding management resulted in allele frequency changes toward 0.5 giving a low rate of increase in homozygosity for the panel used for management, but not for unmanaged neutral loci, at the expense of a high genetic drift. When genomic relationship matrices were based on drift, following VanRaden and as in GCTA, drift was low at the expense of a high rate of increase in homozygosity. The use of IBD-based relationship matrices for inbreeding management limited both drift and the homozygosity-based rate of inbreeding to their target values. Genetic improvement per percent of inbreeding was highest when GOC used IBD-based relationships irrespective of the inbreeding measure used. Genomic relationships based on runs of homozygosity resulted in very high initial improvement per percent of inbreeding, but also in substantial discrepancies between drift and homozygosity-based rates of inbreeding, and resulted in a drift that exceeded its target value. The discrepancy between drift and homozygosity-based rates of inbreeding was caused by a covariance between initial allele frequency and the subsequent change in frequency, which becomes stronger when using data from whole genome sequence.
Management of genetic diversity is usually directed at maintaining the diversity that was present in some population, which serves as a reference point against which diversity in the future is compared. This reference population may be some population in the past or the current population. In the absence of genomic data, the accumulated change in diversity was predicted to be a loss, and could only be described by inbreeding coefficients ( F ) based on pedigree data. These coefficients are the expectations of the loss in genetic variance relative to the reference population in which all alleles are assumed to be drawn at random with replacement, i.e., the classical base population. This description as a loss of variance is strictly for additive traits, but individual allele frequency at a locus among individuals (i.e., 0, ½, 1) is an additive trait. In this perspective, the management of genetic diversity comes down to the management of inbreeding, in particular controlling the rate of inbreeding (Δ F ), or, equivalently, the effective population size: N e = 1/(2Δ F ) ( Falconer and Mackay, 1996 ).
Optimal management of inbreeding in breeding schemes is achieved by optimal contribution (OC) selection ( Meuwissen, 1997 ; Woolliams et al., 2015 ) that, by construction, maximizes the genetic gain made for a given rate of inbreeding. In the era of genomics, Sonesson et al. (2012) concluded that genomic selection requires genomic control of inbreeding, i.e., genomic optimal contribution selection (GOC). With OC, the management of diversity within the population uses the form 1 2 c A ′ c where A is wright’s numerator relationship matrix and c is a set of fractional contributions of candidates to the next generation, and with GOC a genomic relationship matrix G replaces A . This has direct correspondence with the substantial literature on the use of similarity matrices and the fractional contributions of species as measures of species diversity (e.g., Leinster and Cobbold, 2012 ). The similarity matrices in OC use the idea of relationships, which are the scaled (co)variances of breeding values between all pairs of individuals in a population past and present, which links to the wider canon of genetic theory.
In the pre-genomics era, relationships were based on pedigree and pedigree-based coefficients of kinship describing the probability of identity-by-descent (IBD) at neutral loci that are unlinked to any loci under selection. Within this subset of loci, IBD results in a redistribution of genotype frequencies away from Hardy-Weinberg proportions toward homozygosity by p 0 2 ( 1 - F ) + p 0 F , 2 p 0 ( 1 - p 0 ) ( 1 - F ) , and (1 − p 0 ) 2 (1 − F ) + (1 − p 0 ) F for the genotypes AA, Aa and aa, respectively, where p 0 is the original frequency of the A allele ( Falconer and Mackay, 1996 ). This redistribution of genotype frequencies links the changes of heterozygosity [expected to reduce by a factor (1–F)], the within line genetic variance [also reducing by (1–F)], and the genetic drift variance of allele frequencies [ p 0 (1– p 0 )F] to the inbreeding coefficient describing the IBD of sampled alleles. These expected changes do not hold for loci linked to the causal variants of complex traits (QTL), where allele frequencies and genotype frequencies may change non-randomly, and cannot be explained by IBD predicted by pedigree alone.
When defining inbreeding as the correlation between uniting gametes, Wright (1922) assumed the infinitesimal model, which implies infinitesimal selection pressures with random changes in allele frequency. However, the genome is of finite size, and for complex traits with many QTL selection pressures will extend to neutral loci in linkage disequilibrium (LD) across the genome, and these associations to loci under selection result in non-random changes of allele frequencies. This is particularly the case for genomic selection schemes, where marker panels are large, but not infinitely large, dense and genome-wide, and designed to be in LD with all QTL, and where selection is directly for the markers included in the panel. In this setting unlinked neutral loci are likely to be rare, so the classical theory appears redundant.
Despite the apparent loss of a unifying paradigm, genomics opens up a choice of tools that could be used to describe genetic diversity that is wider in scope than the classical genetic variance and inbreeding. For example, tools based on genomic relationships ( VanRaden, 2008 ), runs of homozygosity ( de Cara et al., 2013 ; Luan et al., 2014 ; Rodríguez-Ramilo et al., 2015 ), and linkage analysis ( Fernando and Grossman, 1989 ; Meuwissen et al., 2011 ). Some genomic measures may be better suited for some purposes than others, and so the question arises of what is the purpose of the management of diversity in breeding schemes in addition to what tools to use. Furthermore, when considering tools for genomic inbreeding, there is a need to distinguish which aspect of inbreeding they depict (IBD, heterozygosity/homozygosity, or genetic drift), since in (genomic) selection schemes their expectations may differ from those derived from random allele frequency changes resulting in the genotype frequencies p 0 2 ( 1 - F ) + F p 0 , 2 p 0 ( 1 - p 0 ) ( 1 - F ) , and (1 − p 0 ) 2 (1 − F ) + F (1 − p 0 ).
Most molecular genetic measures of inbreeding are based on the allelic identity of marker loci, and do not directly separate IBD from Identity-By-State (IBS). Genomic relationship matrices which are variants of VanRaden (2008) compensate for this by measuring squared changes in allele frequency relative to a set of reference frequencies. For the purposes of managing changes in diversity relative to the reference population these frequencies would be those relevant to this base generation ( Sonesson et al., 2012 ), although often the frequencies in the current “generation” are used ( Powell et al., 2010 ), or simply the subset of the population for which the genomic data is available; see Legarra (2016) for further discussion on these issues. Providing the base generation is used to define the reference frequencies at neutral unlinked loci ( p 0, k for locus k), the expectation of G VR2 (Method 2; VanRaden, 2008 ) is A , with all loci equally weighted after standardization using the base generation frequencies. In comparison, G VR1 (Method 1) can be viewed as simply re-weighting the loci by 2 p 0, k (1− p 0, k ): i.e., for a single locus, G VR1 and G VR2 yield identical relationship estimates, and extending to many loci G VR 2 uses the simple mean of the single locus estimates whereas G VR 1 uses the weighted mean with 2 p 0, k (1− p 0, k ) as the weights. Extending the argument of Woolliams et al. (2015) for G VR1 , since G VR2 is based on the squares of standardized allele frequency changes, and the management of diversity using G VR2 will constrain these squared standardized changes; this measurement of inbreeding will be denoted as F drift [see Eq. (1B) in Methods section for a more precise definition]. When using 0.5 as the base frequency for all loci, as sometimes proposed, the relationship matrix G VR 0.5 is proportional to homozygosity and molecular coancestry ( Toro et al., 2014 ). Hence, G VR 0.5 may be used to measure homozygosity-based inbreeding, F hom , and the loss of heterozygosity (1– F hom ).
The use of a genomic relationship matrix, G LA , based on linkage analysis for inbreeding management was suggested and studied by Toro et al. (1998) , Wang (2001) , Pong-Wong and Woolliams (2007) , Fernandez et al. (2005) , and Villanueva et al. (2005) . Here the inheritance of the marker alleles is used to determine probabilities of having inheriting the maternal or paternal allele from a parent at the marker loci instead of assuming 50/50 inheritance probabilities as in A . G LA thus requires pedigree and marker information, and IBD relationships are relative to the (assumed) unrelated and non-inbred base population as in A . In this way IBD is evaluated directly by G LA , and is not simply an expectation for neutral unlinked loci as described above for G VR2 . If two (base) individuals are unrelated in A then they are unrelated in G LA , whereas the other measures also estimate (non-zero) relationships for base population individuals. The marker data accounts for Mendelian segregation which may deviate from 50/50 probabilities through any linkage drag from loci under selection, or selective advantage. G LA can be constructed by a tabular method, similar to that for the pedigree based relationship matrix ( Fernando and Grossman, 1989 ), and software for the simultaneous linkage analysis of an entire chromosome is available (e.g., LDMIP (Linkage Disequilibrium Multilocus Iterative Peeling); Meuwissen and Goddard, 2010 ). G LA is a tool that specifically describes IBD across the genome, hence we will denote this IBD based estimate of inbreeding as F IBD .
A run of homozygosity (ROH) is an uninterrupted sequence of homozygous markers ( McQuillan et al., 2008 ). The exact definition of a ROH differs among studies as a number of ancillary constraints are added related to the minimum length of a ROH measured in markers and/or cM, minimum marker density, and in some cases an allowance for some heterozygous genotypes arising from genotyping errors. The idea is that a run of homozygous markers indicates an IBD segment, since it is unlikely that many consecutive homozygous markers are IBS by chance alone. The total length of ROH relative to the total genome length provides an estimate of F IBD from the DNA itself, and this estimate will be denoted F ROH . The reference population for F ROH is unclear, although by varying the constraint on the length of the ROHs the emphasis can be changed from old inbreeding, with short ROHs, to young inbreeding, with long ROHs ( Keller et al., 2011 ). F ROH may miss some relevant inbreeding since IBD segments shorter than the minimum length are neglected. On the one hand, F ROH is an IBD based measure of inbreeding, as it attempts to identify IBD segments (especially when ROHs are long), but on the other hand it is a homozygosity based measure of inbreeding since it is actually based on the homozygosity of haplotypes (especially when ROHs are short). However, F ROH is a measure of inbreeding in a single individual and is unsuitable for a measure of IBD within the population as a whole. Therefore integration of ROH into a GOC framework requires a pairwise measurement to form a similarity matrix, G ROH ( de Cara et al., 2013 ).
The aim of this study is to: (i) re-examine the goals of the management of genetic diversity in breeding schemes, and the molecular genetic parameters that may be incorporated into these goals; and (ii) compare alternative genomic- and pedigree-based measures of inbreeding and relationships for addressing the goals. In doing so the different tools discussed above and some novel variants will be compared for their ability to generate gain in breeding schemes while measures of inbreeding are constrained. Finally, conclusions are made with respect to the practical implementation of these tools for managing diversity and how the outcomes will depend on whether whole genome sequence (WGS) data is considered or marker panels.
Materials and Methods
The goals of the management of genetic diversity.
Managed populations, such as livestock, will generally have many desirable characteristics (related to production, reproduction, disease resistance, etc.). Some of these characteristics are to be improved (the breeding goal traits), without jeopardizing the others. The latter is the aim of the management of inbreeding. Specifically, breeding programs aim to change allele frequencies at the QTL in the desired direction. This ultimately results in loss of variation at the QTL as fixation approaches, but providing these changes are in the right direction this loss of variation is not a problem. However, genetic drift from our reference population and loss of variation at loci that are neutral for the selection goal are to be avoided for the following reasons. Firstly, to alleviate the risk of inbreeding depression through decreased heterozygosity, particularly for traits that are not under artificial selection but are needed for the healthy functioning of the animals. Secondly, deleterious recessive alleles may drift to high frequencies, and occur more frequently in their deleterious or lethal homozygous form; although mentioned separately this is a specific manifestation of inbreeding depression. In the genomics era, deleterious recessives may be identified and mapped ( Charlier et al., 2008 ), and if achieved recessive mutations may be selected against (at the cost of selection pressures), or potentially gene-edited. Nonetheless, simultaneous selection against many genetic defects diverts substantial selection pressures away from other traits in the breeding goal. Thirdly, loss of variation arising from selection sweeps for the current goal may erase variation for traits that are currently not of interest but may be valued in the future and so limit the future selection opportunities. Fourthly, genetic drift in the sense of random changes of allele frequencies, and thus random changes of trait values, which may be deleterious. This encompasses both the traits outside the current breeding goal and within it, where drift is observed as variability in the selection response. Moreover, large random changes in allele frequency may disrupt positive additive-by-additive interactions between QTL which have occurred due to many generations of natural and/or artificial selection (similar to recombination losses in crossbreeding; Kinghorn, 1980 ). In addition, random allele frequency changes may result in the loss of rare alleles, which implies a permanent loss of variation.
Measures for Management of Inbreeding
Whilst genomics offers molecular measures for direct monitoring, most obviously heterozygosity and frequency changes measured from a panel of anonymous markers, the strategy for management of these diverse problems using genomics does not follow directly. For example, increasing heterozygosity per se , achieved by moving allele frequencies of marker loci toward ½ is not solely beneficial, as while potentially ameliorating the aforementioned problems 1 and 3 it is deleterious for problems 2 and 4. Both these empirical measures of heterozygosity and the change of frequencies from drift can be considered to be measures of inbreeding and diversity. Wright (1922) states that a natural inbreeding coefficient moves between 0 and 1 as heterozygosity with random mating moves between its initial state and 0: therefore, if a locus k has initial frequency p 0 and current frequency p t,k then a measure of inbreeding is 1−( H t , k / H 0, k ) = 1−[2 p t , k (1− p t , k )]/[2 p 0, k (1− p 0, k )], which can be generalized by averaging loci to obtain F hom , i.e.,
where N SNP is the total number of loci. F hom can be negative when heterozygosity increases due to allele frequencies moving toward 0.5. Similarly, drift can be measured as δ p t , k 2 = ( p t , k - p 0 , k ) 2 , scaled by the expected value for complete random inbreeding, i.e., δ p t , k 2 / [ p 0 , k ( 1 - p 0 , k ) ] , and similarly averaged over loci to obtain F drift , i.e.,
and which is never negative. F drift is similar to the definition of F ST ( Holsinger and Weir, 2009 ), which is here applied to a single population over time instead of a sample of populations, and it is this empirical measure that is being directly addressed when using G V R 2 .
For locus k in the set of neutral loci with frequency p 0, k in the base population and frequency p t , k = p 0, k + δ p t , k in generation t, twice the frequency in generation t is 2 p t , k 2 + H t , k = 2 ( p 0 + δ p t , k ) , where H t , k = 2( p 0 + δ p t , k )(1− p 0 −δ p t , k ), which holds for all loci assuming random mating. With a sufficiently large subset of neutral loci with the same base frequency p 0 if E [δ p t , k | p 0 ] = 0 then taking expectations over this subset 2 E [ p t , k 2 ] + E [ H t , k ] = 2 p 0 and so 2 ( E [ p t , k 2 ] - p 0 2 ) + E [ H t , k ] = 2 p 0 ( 1 - p 0 ) . The first term is 2 v a r ( p t , k ) and the second is H t and dividing through by 2 p 0 (1− p 0 ) gives
Therefore if E [δ p t , k | p 0 ] = 0 over the range 0 < p 0 < 1, there is an equivalence of F drift with F hom irrespective of initial frequency, p 0 ( Falconer and Mackay, 1996 ): i.e., drift- and homozygosity-based inbreeding are expected to be the same if allele frequency changes are on average 0 irrespective of the initial frequency.
Using a form of GOC related to G VR1 (see Discussion), de Beukelaer et al. (2017) explore the management of diversity and derived the consequences for the rate of homozygosity, 2 ( δ p t , k 2 + 2 δ p t , k ( p 0 - 1 2 ) ) / H t , k . They suggested (supported by results below) that the term δ p t , k ( p 0 - 1 2 ) , which represents a covariance between allele frequency change δ p t , k and initial frequency p 0, k across the loci k , may be non-zero. Consequently, E [δ p t , k | p 0 ]≠0, and Equation [2] will no longer hold, and F drift ≠ F hom . Supplementary Information 1 shows that any deviation from Equation [2] for a general set of loci for which E [δ p t , k ] = 0 over the set, not necessarily with the same initial frequency, must be explained by a covariance between allele frequency changes and the original frequency cov(δ p t , k ; p 0, k ) and shows:
i.e., if there is covariance between initial allele frequencies and frequency changes, homozygosity and drift based inbreeding are no longer equal. Therefore this covariance will be important in determining the impact of genomic management, which aims to manage both the increase of homozygosity and genetic drift.
Supplementary Information 1 explores why completely random selection of parents (i.e., with no management) generates no covariance and how different broad management goals for diversity may generate a covariances of different signs. In particular, with completely random selection, most markers drift to the nearest extreme with the smaller change in frequency, but a minority will move to the opposite extreme resulting in the larger frequency change, giving a net result of no covariance. The consequence of using GOC based on G VR2 is that the latter large allele frequency changes are penalized more heavily, since they add as δ p t , k 2 to the elements of G VR2 and consequently to 1 2 c G ′ c . Hence, the hypothesis is tested below that G VR2 emphasizes the movement of MAF toward 0, and more generally allele frequencies move away from intermediate values toward the nearest extreme, resulting in c o v (δ p t , k ; p 0, k ) > 0 and v a r ( p t , k )/[ p 0 (1− p 0 )] + E [ H t , k / H 0, k ] < 1, contrary to expectations in Eq. (2).
Conversely if G 0.5 is used in GOC then there will be pressure to move allele-frequencies toward 0.5 resulting in increasing heterozygosity ( Li and Horvitz, 1953 ). Supplementary Information 1 shows that this results in c o v (δ p t , k ; p 0, k ) < 0, and thus F hom < 0, and F drift > 0, and v a r ( p t , k )/[ p 0 (1− p 0 )] + E [ H t , k / H 0, k ] > 1, again contrary to expectations in Eq. (2). Furthermore the implication of these considerations is that the covariance c o v (δ p t , k ; p 0, k ) is a property of the active management of diversity using squared frequency changes as in G VR2 (or G VR1 ) and not as a consequence of directional selection. This hypothesis was tested below in two ways: firstly by combining the management of diversity using G VR2 with randomly generated EBVs, and secondly by using a panel of markers for managing diversity that is distinct from the panel used for estimating GEBVs for genomic selection.
The term δ p t , k 2 / [ p 0 , k ( 1 - p 0 , k ) ] appearing in F drift can be viewed as an approximation to the squared total intensity ( i 2 ) applied to the marker, where i ≈δ p t , k /[ p 0, k (1− p 0, k )]. The approximation arises because the total selection intensity applied to a marker is not linear with frequency (see Liu and Woolliams, 2010 ). For example, after the initial generation, the intensity applied to alleles moved toward ½ is overestimated, since the denominator of i increases over time, which reduces the actual intensity applied. The opposite holds for those alleles moved toward the nearest extreme. Therefore a further hypothesis is that a relationship matrix built upon i 2 , G i(p) , rather than δ p t , k 2 may remove the covariance of the change in frequency with the initial frequency that is generated using G VR2 . More details on this and the calculation of G i(p) are given in Supplementary Information 2 .
In classical theory, the equivalence of F drift with F hom under random mating is an outcome of considering IBD, and management by IBD. The genomic relationship matrices based on allele frequency changes or functions of these changes no longer consider IBD as they only consider IBS. Supplementary Information 3 considers the IBD properties of the linkage analysis relationship matrix G LA which is derived from the markers. Considering the management of diversity over generations when using G LA , the conclusion of Supplementary Information 3 is that δ p t , k will now be determined by the properties of the base population and not through linkage disequilibrium generated in the course of the selection process. Therefore, the covariance between the change in frequency and its initial value is potentially avoided. This leads to a further hypothesis tested below that if G LA replaces G VR2 in GOC then F drift = F hom and v a r ( p t , k )/[ p 0 (1− p 0 )] + E [ H t , k / H 0, k ] = 1, as expected in Eq. (2); i.e., consideration of IBD restores the equivalence of F drift and F hom for a set of neutral markers. If A or a ROH-based G ROH replaces G LA the same hypothesis may be advanced given their focus on approximating IBD, however, both are approximations to the true genomic IBD that is tracked by G LA and so the equivalence may only be approximate.
In summary, there are a range of hypotheses to be tested on three categories of relationship matrix: those based on drift, changes in allele frequency or functions of them ( G V R 1 , G V R 2 ,and G i ( p ) ); those based on homozygosity exemplified by G 0.5 ; and those based on IBD ( G LA and A ). A relationship matrix based on ROH, G ROH , is a hybrid of the latter two, targeting IBD by measuring homozygosity of haplotypes.
Breeding Structure and Genomic Architecture
A computer simulation study was conducted to compare these alternative GOC methods. The simulations mimicked a breeding scheme using sib-testing, such as those used for disease challenges in fish breeding, which is similar to Sonesson et al. (2012) . The scheme had a nucleus where selection of candidates was entirely based on their genomic data and performance recording was solely on the full-sibs of the selection candidates which were also genotyped. This scheme may be considered extreme in the sense that the candidates themselves have no performance records, and is practiced in aquaculture to prevent disease infections within the breeding population. There were 2000 young fish per generation, and every full-sib family was split in two: half of the sibs became selection candidates and the other half test-sibs. The actual number of families and their size depended on the optimal contributions of the parents.
The genome consisted of 10 chromosomes of size 1 Morgan. Base population genomes were simulated for a population of an effective size of N e = 100 for 400 (=4 N e ) generations with SNP mutations occurring at a rate of 10 –8 per base pair per generation using the infinite-sites model. This resulted in WGS data for base population genomes that were in mutation-drift-linkage disequilibrium balance. The historical population size was chosen to equal the effective population size targeted for the breeding schemes and so avoid any effect of a sudden large change in effective population size. This resulted in 33,129 segregating SNP loci, which is relatively small in number due to the small effective size of 100. From these loci N SNP = 7000 were randomly sampled as marker loci for use in obtaining GEBV by genomic selection (Panel M); another distinct sample of 7000 loci were randomly sampled as additive QTL, which obtained an allelic effect sampled from the Normal distribution (Panel Q); and a further distinct sample of 7000 SNP loci were randomly sampled to act as “neutral loci” (Panel N), which were used to assess allele-frequency changes and loss of heterozygosity at neutral (anonymous) WGS loci, not involved in either genomic prediction or diversity management. In the majority of schemes Panel M was used for constructing genomic relationship matrices for both obtaining EBVs and diversity management. However, to test whether the non-neutrality of the SNPs used for genomic prediction interfered with their simultaneous use for diversity management, a further distinct panel of 7000 randomly picked loci (Panel D) was used for diversity management in some schemes.
True breeding values were obtained by summing the effects of the QTL alleles across the loci in Panel Q, before scaling them such that the total genetic variance was σ g 2 = 1 in the base population. Phenotypes were obtained by adding a randomly sampled environmental effect with variance σ e 2 = 1.5 , resulting in a heritability of 0.4. After the initial 400 unselected generations to simulate a base population ( t = 0), the breeding schemes described below were run for 20 generations, of which the first generation comprised random selection in order to create an initial sib-family structure.
Genomic Estimates of Breeding Values
GEBV ( g ^ ) were obtained by the SNP-BLUP method ( Meuwissen et al., 2001 ) where BLUP estimates of SNP effects were obtained from random regression on the SNP genotypes of Panel M coded as X ik = –2 p 0, k /√[2 p 0, k (1– p 0, k )], (1–2 p 0, k )/√[2 p 0, k (1– p 0, k )], or (2–2 p 0, k )/√[2 p 0, k (1– p 0, k )] for homozygote, heterozygote, and alternative homozygote genotypes, respectively, of the k th SNP of animal i , and p 0, k is the allele frequency of a randomly chosen reference allele of the k th SNP in generation 0. The model for the BLUP estimation of the SNP effects was:
where y is a vector of records; μ is the overall mean; X is a matrix of genotype codes as described above; b is a vector of random SNP effects [ a priori , b ∼ M V N ( 0 , σ g 2 N S N P - 1 I ) ], and e is a vector of random residuals [ a priori e ∼ N ( 0 , σ e 2 I ) ]. GEBV were obtained as g ^ = X b ^ where b ^ denotes the BLUP estimates of the SNP effects. This model is often implemented in the form of GBLUP using VanRaden (2008) Model 2, which assumes that all loci explain an equal proportion of the genetic variance. When simulating true breeding values, variances of allelic effects were equal across the loci, which implies that the high-MAF QTL explain more variance than the low-MAF QTL. Hence, there is a discrepancy between the simulation model and that used for analysis. However, such discrepancies always occur with real data. To separate the effects of selection and inbreeding management, one of the schemes described below randomly sampled GEBVs from a Normal distribution each generation.
Assessing the Rates of Inbreeding at Neutral Loci
F hom and F drift were calculated for each scheme, and since discrepancies were anticipated ( Supplementary Information 1 ) Δ F was also calculated from both heterozygosity and drift to give Δ F hom and Δ F drift . The calculations described below were done for all schemes with Panel N which were both functionally neutral in not influencing the breeding goal traits, and algorithmically neutral in not being involved in the breeding value prediction. Calculations were repeated for Panel M, and Panel D when used.
Heterozygosity
Calculation was based upon classical models where for generation t (Σ loci k H t , k / H 0, k )/ N SNP = 1− F hom = (1−Δ F ) t where Δ F is the rate of inbreeding, and N SNP the number of loci in the panel. A log transformation yields a linear relationship log(Σ loci k H t , k / H 0, k )−log( N SNP ) = t log(1−Δ F )≈− t Δ F , where the approximation holds for small Δ F when using natural logarithms. This regression was calculated and provided both a test of constant Δ F hom and an estimate of Δ F hom from (−1) × slope of the regression.
At time t , F drift was calculated as Σ loci k ( p t , k − p 0, k ) 2 /[ p 0, k (1− p 0, k )]. Analogously with heterozygosity, classical theory was followed by taking logs of (1− F drift ) with Δ F drift estimated by −1 × slope from the regression on t .
Optimum Contribution Selection Methods
In optimal contribution selection, the rate of inbreeding is constrained by constraining the increase of the group coancestry of the selected parents, G ¯ = 1 2 c ′ G c , where G denotes the relationship matrix of interest for managing diversity among the selection candidates, and c denotes a vector of contributions of the selection candidates to the next generation, which is proportional to their numbers of offspring. Therefore the group coancestry is the average relationship among all pairs of the parents, including self-pairings, weighted by the fraction of offspring from the pair assuming completely random mating. Furthermore, the genetic level of the selected animals, g ¯ = c ′ g ^ , is maximized weighted by their number of offspring. Hence, the optimisation is as follows:
A number of relationship matrices were investigated for managing the diversity: (i) the pedigree-based relationship matrix A ; (ii) the genomic relationship matrix G VR 2 = X X ′/ N SNP ( VanRaden, 2008 ; Model 2) constructed using Panel M; (iii) the genomic relationship matrix G V R 1 = Z Z ′/Σ loci k H 0, k ( VanRaden, 2008 ; Model 1) constructed using SNP Panel M where Z i j = (−2 p 0 j ),(1−2 p 0 j ),or(2−2 p 0 j ); (iv) G 0.5 , a homozygosity based matrix of relationships, since its elements ( i,j ) are proportional to the expected homozygosity of progeny of animals i and j ( Toro et al., 2014 ); (v) G LA constructed from Panel M using linkage analysis ( Fernando and Grossman, 1989 ; Meuwissen et al., 2011 ); (vi) a novel relationship matrix G i(p) constructed from squared total applied intensities using Panel M (see Supplementary Information 2 ); (vii) the genomic relationship matrix G ROH based on ROH assessed using Panel M following the method of de Cara et al. (2013) (see Supplementary Information 2 ); (viii) a genomic relationship matrix G VR2 constructed using Panel D instead of M. In this replicated simulation study, the calculation of G LA by LDMIP ( Meuwissen and Goddard, 2010 ) was computationally too demanding and instead, a haplotype-based approach was adopted as an approximation (see Supplementary Information 2 ).
Implementation of Selection Procedures
The selection schemes simulated will be denoted by the relationship matrix used in GOC and the panel of markers used for SNP-BLUP and building the relationship matrix. The panel for SNP-BLUP was either “M”, or “∼” when using randomly generated GEBV. The latter implements a scheme without directional selection, and tests whether observed results are due to selection or due to diversity management. The panel for management of inbreeding was either “M,” “D,” or “∼” when using A which required no marker panel. Therefore a total of 9 schemes contribute to the results presented: 6 of which are of the form G (M,M) where G is either G VR 1 , G VR2 , G 0.5 , G LA , G i(p) , and G ROH ; with the remaining three being A (M,∼), G VR2 (M,D), and G VR2 (∼,M), where the first symbol in parentheses refers to EBV estimation and the second to diversity management. The schemes are summarized in Table 1 .

Table 1. The relationship matrices and marker panels that were used for the alternative breeding schemes.
For all schemes the target Δ F was set via the parameter K to 0.005 / generation, so the target effective population size was 100. Therefore the group coancestry of the parents was set in generation t to K t = K t −1 + 0.005(1− K t −1 ), where K 0 = 1 / 2 G ¯ and G ¯ denotes the average relationship of all candidates in generation 1 (the first generation with GOC selection). Each scheme was replicated 100 times by generating a new base population as described above. Simulation errors were reduced by simulating all alternative breeding schemes on each replicate of the initial generations, using the same Panels M, Q, N, and D, and the same effects for the QTLs. Each generation had random mating among males and females with mating proportions guided by the optimum contributions c .
G LA and A are mathematically guaranteed to be positive definite, and G VR 1 , G VR2 , G 0.5 , and G i(p) are guaranteed to be positive semi-definite, i.e., all eigenvalues λ i ≥0, as they are the cross-product of SNP genotype matrices ( X or Z ) with one eigenvalue of zero due to the centring of the genotypes. For the semi-definite matrices a small value (α = 0.01) was added to their leading diagonal to make them invertible, and positive definite to permit the use of the optimal contribution algorithm of Meuwissen (1997) . In contrast, G ROH is not guaranteed to be semi-positive definite since its elements are calculated one by one, and large negative eigenvalues for G ROH were observed empirically (results not shown). When using a general matrix inversion routine the achieved Δ F were much larger than 0.005/generation. Hence, G ROH was made positive definite by adding substantial values of α to its diagonals, chosen by trial and error. Starting from an initial value of α = 0.05, positive definiteness was tested by inversion using Cholesky decomposition, and if it failed then α was doubled if α < 1 or increased by 1 otherwise, until inversion was successful.
The distribution of MAF for the SNPs in the WGS of the founder population ( t = 0) observed in the simulations is depicted in Figure 1 . The four SNP panels, i.e., M, the SNP-BLUP panel, N, the neutral marker panel, Q, the QTL panel, and D, a second marker panel for genetic diversity management, are random samples from the SNPs depicted in Figure 1 . The MAF distribution is typical for that of whole genome sequence data with very many SNPs with rare alleles and relatively few SNPs with intermediate allele frequencies.

Figure 1. Histogram of the minor allele frequencies (MAF) of the SNPs in the whole genome sequence of the founder population ( t = 0) observed in the simulations following 4000 generations of mutation and random selection.
Equivalence of F drift and F hom
Table 2 shows for the alternative breeding schemes the drift- and homozygosity-based rates of inbreeding, together with the deviations F hom – F drift in generation 20. For classical inbreeding theory the expectation is that F hom = F drift = 0.095 for random mating. However, with two sexes there will be deviations which depend on the number of mating parents which are shown in Figure 2 and were approximately equally divided between males and females each generation. This has an impact in decreasing F hom at generation 20 below random mating expectations by approximately 1/(2T) where T is the total number of parents following Robertson (1965) . Therefore at generation 20, there is a classical expectation for F drift to exceed F hom by ∼0.001 for schemes G ROH (M,M) and A (M,∼), through ∼0.005 for G LA (M,M) to ∼0.01 for G VR2 (M,M).

Table 2. Rates of increase of homozygosity (Δ F hom ), drift (Δ F drift ), and the deviation F hom – F drift in generation 20 for different types of diversity measures for Panels M and N.

Figure 2. The total number of selected parents for each generation for different breeding schemes. The total is the number of animals with optimal contributions >0 required to achieve a fractional increase in the OC constraint of 0.005.
The deviations of F hom – F drift from 0 were significant for all the schemes, for both the SNP-BLUP Panel M and the neutral Panel N, and would imply significant deviations from the classical Eq. (2). The deviation F hom – F drift for G LA (M,M) was closest to the classical expectation, and was closer still after accounting for the degree of non-random mating that was present. Among the remaining schemes A (M,∼) most closely aligns to classical expectations. The results based on ROH which attempts to mimic IBD appears more similar to G 0.5 (M,M) which manages homozygosity, where F drift exceeds F hom , although the deviations of the G 0.5 (M,M) scheme are much larger, with F hom − F drift = −0.347 for Panel M which is more than a third of the maximum inbreeding coefficient of 1.
G VR2 (M,M), i.e., a commonly used GOC scheme, showed a large deviation opposite to that for G 0.5 (M,M) with F hom − F drift = 0.147 for Panel M, and 0.053 for Panel N, an excess of loss of heterozygosity relative to drift. Supplementary Information 1 shows this discrepancy must arise due to a covariance between the direction of allele frequency change and initial frequency, with a stronger drift to extremes than would be expected in classical theory. Figure 3 illustrates this covariance for a randomly chosen replicate, and shows the regression line ( P < 0.001); for this replicate the difference F hom − F drift = 0.055 in Panel N, which arose from a correlation of only 0.040. For G VR1 (M,M), which compared to G VR2 (M,M) weights the Panel M loci proportional to 2 p 0, k (1− p 0, k ), this covariance was weaker but was still observed. The result for G VR2 (M,D) showed that if the panel used for managing diversity (D) is distinct from that used for SNP-BLUP (M), the covariance in Panel M became similar to that for Panel N, as it is no longer directly managed for its diversity, and the outcome for the unmanaged neutral Panel N was almost identical to G VR2 (M,M). The hypothesis that the covariance arises solely as a property of the management by G VR2 , rather than as a consequence of the directional selection, was confirmed by the results for G VR2 (∼,M) where F hom still exceeded F drift . Managing the intensity in scheme G i(p) (M,M) did not remove the covariance but, in contrast to the other “drift” schemes, reversed its sign so that F drift exceeded F hom , which is in accord with the hypothesis that it introduces an increased “cost” of moving toward the extremes compared to G VR2 (M,M).

Figure 3. The covariance between the standardized change in allele frequency at t = 20 and the standardize frequency at t = 0 for the 7000 SNP loci in Panel N for a randomly chosen replicate. Standardization is by p 0 , k ( 1 - p 0 , k ) for locus k . The solid black line is the fitted linear regression y = 0.0083 + 0.0070×, with SES 0.0042 and 0.0021, respectively, and a Pearson correlation r = 0.040. For this replicate F drift = 0.123, F hom = 0.178, and twice the covariance was 0.0555. The upper x -axis shows the untransformed frequency.
Managing the Rates of Inbreeding
Table 2 shows Δ F drift and Δ F hom for the different schemes for Panels M and N, and Figure 4 shows F drift and F hom over time. Figure 4 shows that log(1- F drift ) is approximately linear with generation for all schemes, in contrast to log(1- F hom ) where some schemes, e.g., G ROH (M,M) show marked curvilinearity.

Figure 4. Changes in inbreeding coefficients F drift and F hom for the neutral loci of Panel N over time plotted on a logarithmic scale where a constant rate of inbreeding results in a linear increase of over time: (A) natural logarithm of (1–F hom ); and (B) natural logarithm of (1–F drift ).
For G VR2 (M,M), Δ F drift for Panel M was directly controlled and was on target at 0.005, but Δ F hom was more than double this target, due to the covariance described above. For Panel N, Δ F drift was greater and Δ F hom was less than observed for Panel M, so the difference was less extreme. The increase in Δ F drift was due to Panel N’s LD with QTL that was not accounted for by its LD with Panel M, while the decrease in Δ F hom was due to the allele frequencies for loci in Panel N being subject to weaker regulation due to their imperfect LD with those in Panel M. The same pattern of differences between Δ F drift and Δ F hom was observed in a less extreme form with G VR2 (∼,M) as here the imperfect LD between Panels M and N is still important but the more favored marker alleles in Panel M change randomly from generation to generation. The outcome for Δ F drift shown in Table 2 for G VR1 (M,M) for Panel M is greater than the target, as F drift and F hom weight all loci in a panel equally, whereas the management weights the drift by 2 p 0, k (1− p 0, k ), consequently the LD with QTL is more weakly constrained for loci with low MAF in Panel M, which is where the impact of the covariance is greatest ( Figure 3 ). This also explains the lower Δ F hom observed for G VR1 (M,M). The results for G i(p) (M,M) shown in Table 2 reflect the changed sign in the covariance in that Δ F hom was less than Δ F drift . Unlike G VR2 (M,M), the constraint applied was only indirectly related to F drift or F hom and so the achieved rates were not expected to meet the target, although Δ F hom was close to the target for Panel M.
As with G i(p) (M,M) the simulated management for the measures based on homozygosity, G 0.5 (M,M) and G ROH (M,M), did not explicitly control F drift or F hom , However, Δ F hom was close to the desired target for G ROH (M,M) when measured in both Panels M and N. G ROH (M,M) showed a curvilinear time trend for F hom mainly due to a negative Δ F hom during the first few generations, after which it increased with time and was rising faster than G LA (M,M) at the end of the period; in contrast Δ F drift was approximately linear. The accelerating Δ F hom maybe caused by ROHs failing to accumulate inbreeding as haplotypes recombine, so reducing the length of IBD segments below the thresholds implicit in ROH methods, while this older inbreeding is captured by F hom . To test this, the minimum length of a contributing ROH was halved to ∼3.5 from ∼7 Mb but results were nearly identical to those shown in Table 3 (result not shown). G 0.5 (M,M) has the highest F drift , because it explicitly promotes allele frequency changes to intermediate frequencies for all loci.

Table 3. Genetic gain (and its SE) after 20 generations of selection expressed in initial genetic standard deviation units, and inbreeding measured by homozygosity for Panel N of neutral loci at generation 20 for comparison.
In contrast to all other schemes, Δ F drift for G LA (M,M) was within 2% of the target for both Panels M and N (see Table 2 ) but was below target for Δ F hom for both panels. The discrepancy for Δ F hom is complicated by the dynamic pattern of the number of parents selected in this scheme (see Figure 2 ), which results in the expected heterozygosity being close to that for random mating in early generations, but ∼0.005 less than random mating in later generation as a result of the degree of non-random mating introduced by the smaller number of parents. Therefore estimating Δ F hom from observed heterozygosity will underestimate the true value and explains a substantial part of the observed deviation from the target value of 0.005. Figure 4 shows G LA (M,M) was lowest for F drift and F hom in generation 20 with near constant rates. The results from AOC were qualitatively similar except that both Δ F hom and Δ F drift exceeded the target rates by 40% in both panels. This is due to the hitch-hiking of neutral loci with the changes in QTL frequencies arising from the LD generated within families and is unaccounted by using expectations of IBD based on pedigree.
Genetic Gain
Table 3 shows the genetic gains of the schemes achieved after 20 generations of selection and Figure 5 shows the gain achieved over time as a function of F drift and F hom for the neutral markers in Panel N. Figure 5 allows comparisons to be made at the same F drift or F hom and offsets, in part, the unequal rates of inbreeding observed among the different schemes.

Figure 5. Genetic gain, Gt plotted against inbreeding for generations 1–20, where inbreeding is transformed to a logarithmic scale by –log(1- F t ) for F hom (A) or F drift (B) . For ΔF = 0.005, the target after 20 generations is shown (–log(1- F t ) = 0.1).
The genetic gains were very similar (within 0.3%) for the schemes G VR2 (M,M) and G VR2 (M,D) where the latter differs only in using a second marker panel for inbreeding management which was unambiguously neutral. Given the small difference in their inbreeding rate at the neutral loci in Panel N ( Tables 2 , 3 ), this indicates that separate panels of markers for gain and for diversity is unnecessary for such schemes. The G LA (M,M) scheme yielded significantly more genetic gain than G VR2 (M,M), at lower F drift and F hom . G ROH (M,M) and A (M,∼) yielded substantially more gain, but their F drift was also higher. The A (M,∼) scheme yielded the highest genetic gain of all the schemes compared, but, compared to its closest competitors, G LA (M,M) and G ROH (M,M), it also yielded more F drift and/or F hom .
It is clear from Figure 5 that the ranking of the schemes for achieved gain differs according to whether drift or homozygosity is considered: e.g., G ROH (M,M) and G i(p) (M,M) schemes yielded relatively high gains given F hom , but relatively low gains given F drift , whereas G VR2 (M,M) schemes yielded opposite results with low gains for F hom and relatively high for F drift . The gain for the G ROH (M,M) scheme in early generations was accompanied by negative F hom ( Figure 5A ). G LA (M,M) and A (M,∼) schemes performed relatively well as shown in both plots of Figure 5 , with G LA (M,M) schemes seeming to yield in both plots slightly more gain per unit of inbreeding than A (M,∼). Although, the A (M,∼) gain is high relative to its inbreeding, the inbreeding rates were substantially larger than the target rate (which can be seen from Figure 5 by the curves extending far beyond the target). The G LA (M,M) scheme achieves the target rate of inbreeding closely for Δ F hom and Δ F drift ( Table 2 ), and simultaneously converts inbreeding efficiently into genetic gain. Moreover, when testing genetic gains in generation 20 of the G LA (M,M) schemes to interpolated gains at the same overall inbreeding (average of F hom and F drift ) of the A (M,∼) and G ROH (M,M) schemes, the G LA (M,M) scheme yielded the highest gain in 65, respectively, 62 out of 100 replicates; i.e., generation 20 gains of G LA (M,M) were significantly higher than those of A (M,∼) and G ROH (M,M) ( P < 0.01) at the same averaged inbreeding level.
Number of Parents
Figure 2 shows the number of selected parents across the generations and shows that the schemes that use IBD based relationship matrices ( A , G LA ) and G ROH select most parents. The selected number of parents for G ROH (M,M) may be artificially large due to the additions to the leading diagonal of G ROH (on average 8.7) to make it positive definite. This process made the G ROH matrix diagonally dominant, and so reducing c ’ G ROH c is driven by selecting more parents in order to reduce the impact of these diagonal elements and not about avoiding the selection of related animals. Non-positive definite G ROH matrices could be inverted to obtain optimal solutions c , but these yielded much too high rates of inbreeding (result not shown) probably because optimal contributions c were found that resulted in negative c ’ G ROH c , which does not make sense and inbreeding was high and positive. Schemes using matrices constructed by the methodology of VanRaden (2008) ( G VR1 , G VR2 , G i(p) , and G 0.5 ) select fewest parents, implying that they are able to select relatively less related parents by their respective measure, and differences in relationships are relatively large in their respective matrices. Comparing results from Table 2 and Figure 2 suggests that the selection of relatively few parents is achieved by making use of the opportunities to induce covariances between allele-frequency-changes and initial frequencies that these schemes offer, which in turn affect the frequencies of heterozygotes.
Genetic Variance
Figure 6 shows the genetic variance for the trait calculated from the true breeding values of the individuals. The G 0.5 (M,M) scheme loses substantial genetic variance at an early stage, and this relatively low genetic variance is maintained throughout the 20 generations of selection. Therefore striving for allele frequencies of 0.5 at the loci in Panel M does not maintain variation at the QTL in Panel Q, which is in accord with the results for Panel N in Table 2 . The relatively low variance for A( M,∼) at generation 20 is a consequence of it relatively high genetic gain combined with its relative high rates of inbreeding. By generation 20, the G LA (M,M) scheme has lost least genetic variance, due to its rates of inbreeding not exceeding the target, and may explain why the G LA (M,M) scheme is very efficient in turning inbreeding into gain at the end of the selection period ( Figure 5 ).

Figure 6. The trait genetic variance of the individuals plotted over time.
Equivalence of Measures F hom and F drift
In the classical work of Wright (1922) two natural measures of inbreeding were introduced concerned with the extent of drift on the one hand (here represented by F drift and Δ F drift ) and heterozygosity on the other (here represented by F hom and Δ F hom ), and in classical theory with neutral loci unlinked to QTL these perspectives were identical and directly linked to the occurrence of IBD. The results of this study show that these measures of inbreeding can differ substantially in genomic optimum contribution schemes even when there are no QTL in the genome [ G VR2 (∼,M); Table 2 ]. This is because the management in these schemes is commonly directed at the observed homozygosity or drift of the marker loci being monitored. For example, schemes that limit the rate of increase of homozygosity (as represented here by G 0.5 ) induce a negative covariance between the change in allele frequency and the initial frequency, as an excess of minor alleles compared to classical expectations move toward intermediate levels. Conversely schemes managing drift and limiting changes in allele frequency (e.g., using G V R 2 ) induce a positive covariance between change in allele frequency and the initial frequency, as an excess of minor alleles tend to move toward the nearest extreme. Consequently, systematic discrepancies occur between Δ F drift and Δ F hom . These discrepancies are a property of the inbreeding management and not of selection per se , as they were unaffected by whether random GEBVs were used in the scheme or separate panels of SNPs were used for generating GEBV and management of inbreeding. In contrast to the management using the IBS allele frequencies of monitored markers, when IBD was used either via genomics information ( G LA ) or approximately ( A , uninfluenced by markers) the equivalence of Δ F drift and Δ F hom was re-established in the simulations, although not with G ROH which is targeted toward IBD but is based on the homozygosity of haplotypes.
The origin of these covariances between allele frequency changes and initial frequencies can be seen when considering the form of the relationship matrix and is explored in detail in Supplementary Information 1 . The negative covariance arising from G 0.5 explicitly measures allele frequencies as deviations from 0.5, not from the base frequency p 0, k and consequently gains in this measure of diversity (but not necessarily IBD, as discussed later) are obtained by moving frequencies toward 0.5 offsetting any opposing changes prompted by selection objectives. The positive covariance, for example with G V R 2 , arises because drift of an allele to the more distant extreme is more heavily penalized compared to completely random drift as the GOC with G V R 2 is constraining the square of the change. This will inevitably promote shifts to the nearest extreme, and more strongly so as p 0 deviates more from ½. Since G V R 1 is a re-weighting of the loci in G V R 2 by w k /Σ l o c i k w k for locus k , where w k = 2 p 0, k (1− p 0, k ), placing more weight on frequency changes for loci initially closer to ½, it would be expected the discrepancy between F drift and F hom would be less for G V R 1 than G V R 2 as observed in the simulations (see Table 2 and Figure 4 ). Moving to management using the total intensity applied over time ( G i ( p ) ) penalizes deviations that move toward the extremes more heavily than those toward intermediate frequencies (as d i / d p = [ p (1− p )] −1/2 ; Liu and Woolliams, 2010 ), and this changed the sign of the discrepancy although its magnitude was decreased compared to G V R 2 .
G V R 2 , which was used by Sonesson et al. (2012) , controlled Δ F drift and met the target for the panel used (see Table 2 ) but Δ F hom was much greater due to the covariance discussed above. This agreed with the findings of de Beukelaer et al. (2017) , where it was suggested that the covariance between change in frequency and its initial value could be the cause of this. However, these authors also reset the allele frequencies for the reference population in the G VR1 matrix every generation to the current generation frequencies, which implies that changes in allele frequency in each generation are constrained without reference to their accumulated change over earlier generations. In a continuous selection scheme, the allele frequency changes of successive generations are positively correlated; thus, although the variance of the change in allele frequency within a generation may have been on target, the variance of the cumulative allele frequency change over generations will exceed the target value due to these positive correlations, as observed in their study. This distinction in methodology will have affected all findings on GOC in the study of de Beukelaer et al. (2017) .
Sonesson et al. (2012) found that G V R 2 schemes achieved their target rate of inbreeding based on IBD using loci with 2N alleles scattered across the genome. Details of the founder populations used in their study were presented in Sonesson and Meuwissen (2009) , which revealed that their SNP-BLUP marker panel was selected for intermediate frequencies in order to mimic a typical SNP-chip marker panel. This is very different from the SNP-BLUP panel used here which was a random sample of whole genome sequence data, and hence dominated by extreme allele frequencies ( Figure 1 ). The strength of the covariance underlying the discrepancy between F drift and F hom depends on the distribution of ( p 0 - 1 2 ) , and so in Sonesson et al. (2012) any discrepancy would have been much reduced. In the context of the current results, it was most similar to using G V R 1 where the intermediate loci are more heavily weighted. Conclusions from these considerations are (i) that the discrepancies between the different measures of rates of inbreeding are extreme in WGS data, due to their extreme allele frequencies ( Figure 1 ); and (ii) the discrepancies are a property of the panel used to manage diversity and not the remaining loci, as the IBD-alleles used by Sonesson et al. (2012) have low MAF by construction. Hence, for typical SNPs from chips, the discrepancies between F drift and F hom are expected to be present but smaller than those in Table 2 .
Management of Diversity
An important aspect of a tool to manage diversity is that it is predictable in meeting its targets, and this can be examined for the marker panel, for the unmanaged neutral markers, and for F drift and F hom . In this respect, G VRn meets the target but only for F drift and only in the marker panel (i.e., not in the unmanaged panel) whereas G LA meets the target (with only minor deviations) for both F drift and F hom for both panels. All others failed to meet the target rate to a greater or lesser degree and would need to be calibrated, possibly in every generation, to meet the targets set at neutral loci. In practice, this would require as realistic as possible simulations of the practical breeding scheme using the current situation as a starting point.
A key management objective in breeding schemes is the efficient generation of gain from the genetic variance in the objectives, and conserving the variation at the (currently) neutral loci, and here the IBD-related schemes were best when compared to F drift or F hom of neutral loci. On an average of F drift and F hom , G LA was more efficient than G ROH , which gave different rates for Δ F hom and Δ F drift , would require regular calibration, and (in the current implementation following de Cara et al., 2013 ) always required very large number of parents, which in practice would usually demand additional scheme resources. Henryon et al. (2019) observed that using A appeared to be more efficient than using G V R 2 , and this was confirmed here. The differences between schemes using G LA and A were small when plotted against F drift or F hom but the G LA scheme was the only scheme tested here that combined high efficiency with rates of inbreeding close to and not exceeding the target rate of inbreeding of 0.005. This supports the conclusion of Sonesson et al. (2012) that genomic selection requires genomic control.
One consequence of entering the genomics era is that the meaning of diversity and its management in practice is more open to discussion, as the pedigree is no longer the only tool to measure and manage it. For example, the number of polymorphic loci could be used as a measure, which might underpin major concerns over the disappearance of known rare alleles in the scheme. Further, in the pedigree inbreeding framework, the measure used is the fraction of variance that is expected to have been lost from the reference base. In the genomic era, if the measure is simply defined as the genetic variance defined by IBS and maximized, there is scope for increasing diversity by the directional selection of loci toward intermediate frequencies as an objective. These measures have been explored elsewhere (see Howard et al., 2017 for a review). In general, attaching values (e.g., selection index weights) to genetic diversity is a very difficult task (e.g., Brisbane and Gibson, 1994 ; Wray and Goddard, 1994 ; Goddard, 2009 ; Jannink, 2010 ; Howard et al., 2017 ), which becomes especially clear in view of the aforementioned goals of diversity management, where diversity is required at many (hypothetical) traits simultaneously. Breeders have generally more of an idea about their target rate of inbreeding than on what weight to give to a diversity measure. Although the actual choice of the target rate of inbreeding remains somewhat arbitrary, guidelines have been developed over the years ( Woolliams et al., 2015 , for a review).
Here, it is argued that an over-riding objective for many populations such as livestock or zoo populations, beyond the breeding goals that underlie the selection on the EBV, is to manage over time the risks associated with the unmeasured attributes of a reference population (e.g., unrecognized deleterious recessives, drift in desirable holistic qualities, epistatic variance). In this respect, all approaches used in this study refer back directly to the established reference (base) population. As mentioned above, other perspectives may be advanced such as increasing the genetic variance at neutral loci by increasing heterozygosity (e.g., de Beukelaer et al., 2017 ). This could be achieved by the promotion of allele frequency changes toward intermediate values, as exemplified by G 0.5 in this study, however, this raises issues that require further consideration. Firstly, changes in allele frequency result from multiple copies of a subset of base generation alleles, so increasing frequency is promoting IBD based inbreeding (it is analogous to changing QTL frequency). Secondly, if carried out with a marker panel, then increasing heterozygosity of the marker loci does not necessarily increase heterozygosity among unmonitored neutral loci, which is the objective. In these simulations, the near avoidance of overall loss of heterozygosity in the marker panel by GOC 0.5 during selection was accompanied by much greater drift and more loss of heterozygosity in the unmonitored neutral loci than was achieved using IBD based inbreeding management. In contrast, the use of IBD in G LA has information on the unobserved heterozygosity and drift across all the unmonitored genome positions. It remains only a hypothesis that the management of heterozygosity and drift using IBS might perform better than IBD when WGS sequence data is available, with or without selection, although some studies have considered its use ( Eynard et al., 2015 , 2016 ; Gómez-Romano et al., 2016 ). The question how to weigh F hom and F drift across all loci in the genome when a key objective is to manage unknown or unmonitored risks remains open.
While this study has focused on schemes where loss of genetic diversity is managed next to the maximization of genetic gain, other schemes may be pure conservation schemes, where no genetic change (gain) is desired, but the goals for genetic management are the same; i.e., conserve genetic variation, avoid inbreeding depression, avoid the occurrence of recessive diseases, and avoid random changes in phenotypic traits related to drift from a valued reference population. Strictly, with pure random selection, drift and homozygosity based inbreeding are expected to be the same [Eq. (2); and Falconer and Mackay, 1996 ]. However, minimisation of allele frequency changes or minimisation of loss of heterozygosity based on using IBS may still result in discrepancies between drift and homozygosity based inbreeding measures arising from the covariances described above. In fact, the potential covariance between the change in allele frequency and the initial frequency is expected to increase, since the inbreeding management term is more important in pure conservation schemes. This would also hold for GOC schemes with selection that aim for an N e higher than our goal of N e = 100. The greater potential for discrepancy argues for the use of IBD-based measures of relationship ( G LA , or a more conservative use of A ) to maintain diversity in such genetic conservation schemes.
The approach adopted here has not favored genetic variation at some neutral loci more than others a priori . Of course, a weighted genomic relationship matrix could be implemented and/or the multiple relationship matrices and associated constraints could be used to simultaneously control the genomic variation in different types of loci ( Dagnachew and Meuwissen, 2016 ; Gómez-Romano et al., 2016 ). For example, a general G matrix covering the entire genome, and an additional G matrix controlling genetic diversity at e.g., the major histocompatibility complex, which is essential to the immune response of the animals. Alternatively, regions of the genome may be sought where average heterozygosity is to be increased (reduced) under the assumption that diversity is especially (or not) important in these regions. Regions with known recessive defects may be prioritized for diversity management, but direct inclusion of the known defects in the breeding goal seems more effective in controlling their frequencies. In practice, such regions with special emphasis for diversity management would need to be known a priori , and may only be effective if WGS was used for the relationships because, as shown here, what happens in a sample of loci does not necessarily predict what happens at loci outside that subset. Causative alleles of quantitative traits are quite evenly distributed across the genome ( Wood et al., 2014 ), and as argued here the main goals of diversity management address many anonymous, unknown loci and hypothetical traits simultaneously, which makes it very hard to achieve a worthwhile prioritization of genomic regions for diversity management.
• Contrary to classic inbreeding theory, inbreeding of unmanaged neutral loci as measured by drift ( F drift ) and by homozygosity ( F hom ) can differ very substantially, due to a covariance between the change in allele frequency and its initial frequency, leading to non-zero expected changes in frequency of a sign and magnitude determined by the initial frequency. Discrepancy between F drift and F hom occurs when inbreeding management is based on genomic relationship matrices (or similarity matrices) derived using IBS, but not when derived using IBD, which acts as a unifying concept for F drift and F hom .
• The covariance generated is expected to be larger for WGS data where allele frequencies are extreme with typical MAF close to 0, than for SNP (chip) panels where allele frequencies are generally closer to ½.
• The (genomic) selection component of OC schemes does not cause the difference between F drift and F hom .
• Using the same or a different panel for estimating GEBVs than for management of diversity in OC schemes makes only very small differences to genetic gain and the inbreeding in unmonitored neutral loci.
• Measures of genomic relationship can be classified as those based on changes in allele frequency change (e.g., G VR2 ) and directed at F drift ; those based on homozygosity (e.g., G 0.5 ) and directed at F hom ; and IBD based (e.g., G LA ); or combinations of these (e.g., G ROH ). The choice of the relationship matrix depends very much on what objective it should serve.
• OC schemes that limit F drift directly limit allele frequency changes, such as those using G VR2 , result in low Δ F drift at the expense of high Δ F hom . Schemes using G VR1 will be less extreme in this than G VR2 .
• OC schemes that limit Δ F hom (e.g., using G 0.5 ), result in very low Δ F hom at the expense of high Δ F drift but both F hom and F drift may exceed targets at unmonitored neutral loci.
• The OC scheme using G LA , an IBD based relationship matrix, was the only scheme investigated here that managed homozygosity and drift based inbreeding within the target rate of 0.5%, yielding an effective population size ∼100; for all other schemes, either Δ F drift or Δ F hom or both exceeded their target.
• The OC scheme using G LA yielded the highest gain per unit of inbreeding across both measures of inbreeding, closely followed by the scheme using A . The latter yielded high gain per unit of F but grossly exceeds target rates of inbreeding.
• The use of G LA in practice requires the development of fast algorithms for its calculation.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
TM contributed to study design, performed the simulations, and wrote the draft manuscript. AS developed the simulation software and contributed to discussions and the writing of the manuscript. GG contributed to discussions and the writing of the manuscript. JW contributed to study design, alternative schemes and methods, and discussions and writing of the manuscript. All authors approved the final version of the manuscript.
We are grateful for funding from the Norwegian Research Council (Grant 226275/E40). JW would like to acknowledge funding from the European Commission under Grant Agreement 677353 (IMAGE) and BBSRC Institute Strategic Programe BBS/E/D/30002275.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank three reviewers for their very helpful comments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00880/full#supplementary-material
Brisbane, J. R., and Gibson, J. P. (1994). Balancing selection response and rate of inbreeding by including genetic relationships in selection decisions. World Congr. Genet. Appl. Livest. Prod. 19:135.
Google Scholar
Charlier, C., Coppieters, W., Rollin, F., Desmecht, D., Agerholm, J. S., Cambisano, N., et al. (2008). Highly effective SNP-based association mapping and management of recessive defects in livestock. Nat. Genet. 40, 449–454. doi: 10.1038/ng.96
PubMed Abstract | CrossRef Full Text | Google Scholar
Dagnachew, B. S., and Meuwissen, T. H. E. (2016). A fast iterative algorithm for large scale optimal contribution selection. Gen. Sel. Evol. 48:70.
de Beukelaer, H., Badke, Y., Fack, V., and deMeyer, G. (2017). Moving beyond managing realized genomic relationship in long-term genomic selection. Genetics 206, 1127–1138. doi: 10.1534/genetics.116.194449
de Cara, M. A. R., Villanueva, B., Toro, M. A., and Fernández, J. (2013). Using genomic tools to maintain diversity and fitness in conservation programmes. Mol. Ecol. 22, 6091–6099. doi: 10.1111/mec.12560
Eynard, S. E., Windig, J. J., Hiemstra, S. J., and Calus, M. P. (2016). Whole-genome sequence data uncover loss of genetic diversity due to selection. Genet. Sel. Evol. 48:33.
Eynard, S. E., Windig, J. J., Leroy, G., van Binsbergen, R., and Calus, M. P. (2015). The effect of rare alleles on estimated genomic relationships from whole genome sequence data. BMC Genet. 16:24. doi: 10.1186/s12863-015-0185-0
Falconer, D. S., and Mackay, T. F. C. (1996). Introduction To Quantitative Genetics. Harlow: Pearson Education Limited.
Fernandez, J., Villanueva, B., Pong-Wong, R., and Toro, M. A. (2005). Efficiency of the use of pedigree and molecular marker information in conservation programs. Genetics 170, 1313–1321. doi: 10.1534/genetics.104.037325
Fernando, R. L., and Grossman, M. (1989). Marker assisted selection using best linear unbiased prediction. Gen. Sel. Evol. 21, 467–477.
Goddard, M. E. (2009). Genomic selection: prediction of accuracy and maximisation of long term response. Genetica 136, 245–257. doi: 10.1007/s10709-008-9308-0
Gómez-Romano, F., Villanueva, B., Fernández, J., Woolliams, J. A., and Pong-Wong, R. (2016). The use of genomic coancestry matrices in the optimisation of contributions to maintain genetic diversity at specific regions of the genome. Genet. Sel. Evol. 48:2.
Henryon, M., Liu, H., Berg, P., Su, G., Nielsen, H. M., Gebregewergis, G. T., et al. (2019). Pedigree relationships to control inbreeding in optimum-contribution selection realise more genetic gain than genomic relationships. Genet. Sel. Evol. 51:39.
Holsinger, K. E., and Weir, B. S. (2009). Genetics in geographically structured populations: defining, estimating and interpreting F(ST). Nat. Rev. Genet. 10, 639–650. doi: 10.1038/nrg2611
Howard, J. T., Pryce, J. E., Baes, C., and Maltecca, C. (2017). Invited review: inbreeding in the genomics era: Inbreeding, inbreeding depression, and management of genomic variability. J. Dairy Sci. 100, 6009–6024. doi: 10.3168/jds.2017-12787
Jannink, J. L. (2010). Dynamics of long-term genomic selection. Genet. Sel. Evol. 42:35.
Keller, M. C., Visscher, P. M., and Goddard, M. E. (2011). Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics 189, 237–249. doi: 10.1534/genetics.111.130922
Kinghorn, B. P. (1980). The expression of recombination loss in quantitative traits. J. Anim. Breed. Genet. 97, 138–143. doi: 10.1111/j.1439-0388.1980.tb00919.x
CrossRef Full Text | Google Scholar
Legarra, A. (2016). Comparing estimates of genetic variance across different relationship models. Theor. Popul. Biol. 107, 26–30. doi: 10.1016/j.tpb.2015.08.005
Leinster, T., and Cobbold, C. A. (2012). Measuring diversity: the importance of species similarity. Ecology 93, 477–489. doi: 10.1890/10-2402.1
Li, C. C., and Horvitz, D. G. (1953). Some methods of estimating the inbreeding coefficient. Am. J. Hum. Genet. 5, 107–117.
Liu, A. Y., and Woolliams, J. A. (2010). Continuous approximations for optimizing allele trajectories. Genet. Res. 92, 157–166. doi: 10.1017/s0016672310000145
Luan, T., Yu, X., Dolezal, M., Bagnato, A., and Meuwissen, T. H. (2014). Genomic prediction based on runs of homozygosity. Genet. Sel Evol. 46:64. doi: 10.1016/j.cancergen.2018.04.038
McQuillan, R., Leutenegger, A. L., Abdel-Rahman, R., Franklin, C. S., Pericic, M., Barac-Lauc, L., et al. (2008). Runs of homozygosity in European populations. Am. J. Hum. Genet. 83, 359–372.
Meuwissen, T. H. E. (1997). Maximizing the response of selection with a pre-defined rate of inbreeding. J. Anim. Sci. 75, 934–940.
Meuwissen, T. H. E., and Goddard, M. E. (2010). The use of family relationships and linkage disequilibrium to impute phase and missing genotypes in up to whole-genome sequence density genotypic data. Genetics 185, 1441–1449. doi: 10.1534/genetics.110.113936
Meuwissen, T. H. E., Hayes, B. J., and Goddard, M. E. (2001). Prediction of total genetic value using genome-wide dense marker maps. Genetics 157, 1819–1829.
Meuwissen, T. H. E., Luan, T., and Woolliams, J. A. (2011). The unified approach to the use of genomic and pedigree information in genomic evaluations revisited. J. Anim. Breed. Genet. 128, 429–439. doi: 10.1111/j.1439-0388.2011.00966.x
Pong-Wong, R., and Woolliams, J. A. (2007). Optimisation of contribution of candidate parents to maximise genetic gain and restricting inbreeding using semidefinite programming. Genet. Sel. Evol. 39, 3–25.
Powell, J. E., Visscher, P. M., and Goddard, M. E. (2010). Reconciling the analysis of IBD and IBS in complex trait studies. Nat. Rev. Genet. 11, 800–805. doi: 10.1038/nrg2865
Robertson, A. (1965). The interpretation of genotypic ratios in domestic animal populations. Anim. Prod. 7, 319–324. doi: 10.1017/s0003356100025770
Rodríguez-Ramilo, S. T., Fernández, J., Toro, M. A., Hernández, D., and Villanueva, B. (2015). Genome-wide estimates of coancestry, inbreeding and effective population size in the Spanish Holstein population. PLoS One 10:e0124157. doi: 10.1371/journal.pone.0124157
Sonesson, A. K., and Meuwissen, T. H. E. (2009). Testing strategies for genomic selection in aquaculture breeding programs. Genet. Sel. Evol. 41:37.
Sonesson, A. K., Woolliams, J. A. W., and Meuwissen, T. H. E. (2012). Genomic selection requires genomic control of inbreeding. Genet. Sel. Evol. 44:27.
Toro, M. A., Silio, L., Rodriganez, J., and Rodriguez, C. (1998). The use of molecular markers in conservation programmes of live animals. Genet. Sel. Evol. 30:585. doi: 10.1186/1297-9686-30-6-585
Toro, M. A., Villanueva, B., and Fernandez, J. (2014). Genomics applied to management strategies in conservation programmes. Livestock Sci. 166, 48–53. doi: 10.1016/j.livsci.2014.04.020
VanRaden, P. M. (2008). Efficient methods to compute genomic predictions. J. Dairy Sci. 91, 4414–4423. doi: 10.3168/jds.2007-0980
Villanueva, B., Pong-Wong, R., Fernandez, J., and Toro, M. A. (2005). Benefits from marker-assisted selection under an additive polygenic genetic model. J. Anim. Sci. 83, 1747–1752. doi: 10.2527/2005.8381747x
Wang, J. (2001). Optimal marker-assisted selection to increase the effective size of small populations. Genetics 157, 867–874.
Wood, A. R., Esko, T., Yang, J., Vedantam, S., Pers, T. H., Gustafsson, S., et al. (2014). Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 46, 1173–1186.
Woolliams, J. A., Berg, P., Dagnachew, B. S., and Meuwissen, T. H. E. (2015). Genetic contributions and their optimization. J. Anim. Breed. Genet. 132, 89–99. doi: 10.1111/jbg.12148
Wray, N. R., and Goddard, M. E. (1994). Increasing long term response to selection. Genet. Sel. Evol. 26:431. doi: 10.1186/1297-9686-26-5-431
Wright, S. (1922). Coefficients of inbreeding and relationships. Amer. Nat. 56, 330–338.
Keywords : inbreeding, genetic drift, optimum contribution selection, genetic diversity, genomic relationships, genetic gain
Citation: Meuwissen THE, Sonesson AK, Gebregiwergis G and Woolliams JA (2020) Management of Genetic Diversity in the Era of Genomics. Front. Genet. 11:880. doi: 10.3389/fgene.2020.00880
Received: 16 May 2019; Accepted: 17 July 2020; Published: 13 August 2020.
Reviewed by:
Copyright © 2020 Meuwissen, Sonesson, Gebregiwergis and Woolliams. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY) . The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Theo H. E. Meuwissen, [email protected]
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
- Research article
- Open access
- Published: 07 July 2010
Population genetic diversity and fitness in multiple environments
- Jeffrey A Markert 1 , 2 , 4 ,
- Denise M Champlin 1 ,
- Ruth Gutjahr-Gobell 1 ,
- Jason S Grear 1 ,
- Anne Kuhn 1 ,
- Thomas J McGreevy Jr 1 , 3 ,
- Annette Roth 2 ,
- Mark J Bagley 2 &
- Diane E Nacci 1
BMC Evolutionary Biology volume 10 , Article number: 205 ( 2010 ) Cite this article
20k Accesses
179 Citations
12 Altmetric
Metrics details
When a large number of alleles are lost from a population, increases in individual homozygosity may reduce individual fitness through inbreeding depression. Modest losses of allelic diversity may also negatively impact long-term population viability by reducing the capacity of populations to adapt to altered environments. However, it is not clear how much genetic diversity within populations may be lost before populations are put at significant risk. Development of tools to evaluate this relationship would be a valuable contribution to conservation biology. To address these issues, we have created an experimental system that uses laboratory populations of an estuarine crustacean, Americamysis bahia with experimentally manipulated levels of genetic diversity. We created replicate cultures with five distinct levels of genetic diversity and monitored them for 16 weeks in both permissive (ambient seawater) and stressful conditions (diluted seawater). The relationship between molecular genetic diversity at presumptive neutral loci and population vulnerability was assessed by AFLP analysis.
Populations with very low genetic diversity demonstrated reduced fitness relative to high diversity populations even under permissive conditions. Population performance decreased in the stressful environment for all levels of genetic diversity relative to performance in the permissive environment. Twenty percent of the lowest diversity populations went extinct before the end of the study in permissive conditions, whereas 73% of the low diversity lines went extinct in the stressful environment. All high genetic diversity populations persisted for the duration of the study, although population sizes and reproduction were reduced under stressful environmental conditions. Levels of fitness varied more among replicate low diversity populations than among replicate populations with high genetic diversity. There was a significant correlation between AFLP diversity and population fitness overall; however, AFLP markers performed poorly at detecting modest but consequential losses of genetic diversity. High diversity lines in the stressful environment showed some evidence of relative improvement as the experiment progressed while the low diversity lines did not.
Conclusions
The combined effects of reduced average fitness and increased variability contributed to increased extinction rates for very low diversity populations. More modest losses of genetic diversity resulted in measurable decreases in population fitness; AFLP markers did not always detect these losses. However when AFLP markers indicated lost genetic diversity, these losses were associated with reduced population fitness.
Decreased population genetic diversity can be associated with declines in population fitness (e.g., [ 1 , 2 ]). These declines are thought to involve components of the so called genetic 'extinction vortex', which directly ties losses in population genetic diversity to increased extinction risk [ 3 ]. These losses cause a decrease in individual fitness through the expression of inbreeding depression-like effects, further reducing the effective population size ( N e ) and leading to additional increases in the number of alleles that are alike by state within individuals [ 4 ]. The impact of increased individual homozygosity on individual fitness has been extensively documented in both laboratory, semi-natural, and natural settings [ 2 , 5 – 9 ]. The effects are especially strong in altered or degraded environments [ 10 – 12 ], although the genomic basis of heterozygosity-associated fitness differences and heterosis are still debated [ 13 – 16 ]. In addition to increasing individual homozygosity, lost population genetic diversity also reduces the adaptive potential of a population. For populations to persist over extended time-spans, they must have sufficient allelic resources to adjust to novel selective regimes. Forces ranging from invasive parasites and diseases to shifting climatic patterns ensure that environmental conditions will fluctuate temporally and spatially for all populations. Some species have shown a striking capacity to rapidly adapt to novel selective pressures [ 17 , 18 ] while others have not [ 19 , 20 ]. Because overall population diversity affects both short-term individual fitness and long-term population adaptive capacity, there is a need to develop an empirical quantitative understanding of the relationship between population genetic diversity and population viability.
Many laboratory models have demonstrated the large role of genetic diversity in increasing population fitness mediated through heterosis, particularly when inbreeding levels are high. In one classic example, Leberg [ 21 ] found that populations of mosquito-fish founded with siblings grew more slowly than those founded by unrelated individuals. In a subsequent experiment using non-relatives and experimentally manipulated levels of genetic diversity, Leberg [ 22 ] detected no evidence of a relationship between genetic diversity and population fitness. By manipulating N e while holding the census size constant over three generations in the annual plant Clarkia pulchella , Newman and Pilson [ 23 ] were able to demonstrate that populations with a small N e were more than twice as likely to go extinct as larger populations. Similarly, in a multi-generation experiment using houseflies, Bryant et al. [ 24 ] detected clear declines in relative fitness in low founder number populations and in repeatedly bottlenecked populations, even when the number of individuals subjected to a given bottleneck was relatively large.
Frankham et al. [ 25 ] developed a more direct method for measuring the effect of population genetic diversity on adaptive potential by steadily increasing the level of an environmental stressor (NaCl) every generation in laboratory Drosophila populations. In this study, both mildly bottlenecked and highly inbred populations showed a reduced ability to evolve tolerance to an environmental stressor relative to outbred populations.
In order to understand long-term population viability in a changing environment, experimental models that can build upon these results must be developed. Several published studies provide evidence that severely reduced genetic diversity can affect population fitness, but the impacts on population viability of modest (and perhaps more commonly occurring) reductions in genetic diversity are less well characterized. Further, many laboratory studies of evolutionary processes have relied on Drosophila or Tribolium (e.g., [ 8 , 25 , 26 ]). Both organisms have many experimental advantages, but their very high fecundities [ 27 , 28 ]--which can facilitate rapid rates of adaptation--make them poor models for vertebrate species with much lower reproductive rates. Laboratory models with lower fecundity may be more directly relevant to vertebrate conservation. Ideally, models of evolutionary genetics should also be able to disentangle the effects of population history and the effects of inbreeding from the adaptive potential represented by genetic diversity per se . To do this, they must also allow for fitness to be measured in multiple environments.
Here we present data from laboratory populations of the mysid shrimp ( Americamysis bahia ) a small crustacean native to estuaries along the US East coast [ 29 ]. This animal model has several experimental advantages that make it a valuable tool in evolutionary and conservation genetics. Because they are widely used in toxicological studies, optimal culture conditions and demographics are well characterized [ 30 – 32 ]. Time from conception to first mating is approximately three weeks at 25°C and 30 parts per thousand (ppt) salinity [ 31 , 33 ]. Mature females can produce a new brood every seven days and provide an unusually high level of brood care for an invertebrate; they incubate a small number of fertilized eggs in a marsupium for seven days, giving A. bahia a reproductive profile more similar to many birds and mammals than to other more fecund invertebrates. Owing to their estuarine habitat, A. bahia tolerate a wide range of salinities. In laboratory settings at 25°C, A. bahia cultures reproduce well in natural seawater with a salinity of 31 ppt NaCl, although they are reproductively viable in as little as 10 ppt NaCl [ 31 ]. In the wild, A. bahia have been collected in waters with salinity as low as 3 ppt, although some field surveys suggest they are uncommon below 9 ppt [ 34 ].
By simultaneously manipulating the selective environment and genetic diversity under controlled laboratory conditions with replication, we used A. bahia cultures to develop a more detailed understanding of the relationship between genetic diversity and population fitness in a changing environment. We also generated AFLP [ 35 ] genotypes for many of the populations to determine how well a typical molecular genetic fingerprint analysis predicts meaningful losses of genetic diversity. Our study goal was to develop a model system for quantifying the general relationship between genetic diversity and fitness in both permissive and stressful environments.
Collection of stock populations
Americamysis bahia were collected by dragging a fine-mesh net in shallow waters near Biloxi Beach, MS USA (N30.39351, W088.90123) and Navarre Beach, FL USA (N30.38964, W086.83050) during April 2005. Live animals were keyed out under dissecting microscopes at the US-EPA's Gulf Ecology Division in Gulf Breeze, FL USA. Approximately 50 individuals from each collection site were then transported to the US-EPA's Atlantic Ecology Division facilities in Narragansett, RI USA. Populations derived from each of the two collection sites were housed separately in four 80 L tanks with flow-through seawater maintained at 25°C and an ambient salinity of approximately 30 ppt. Animals were fed Selco enriched Artemia ad lib [ 36 ]. Americamysis bahia cultures grew quickly to more than 2000 individuals from each source.
Generation of low diversity lines
Generation of high diversity lines, salinity and culture.
A pilot study demonstrated that reproductive rates for A. bahia were similar in ambient seawater and at 10 ppt salinity (data not shown). When the salinity was reduced to 7 ppt, reproduction ceased. Based on this preliminary data, together with published findings [ 31 ], and our expectation that low genetic diversity populations would be more sensitive to environmental stress, we chose 9 ppt salinity as the target level of novel environmental stress.
Experimental populations were housed in 9.4 L tanks with precisely controlled salinities, light cycles, and temperatures. Both ambient seawater and seawater diluted with dechlorinated tap water were available via a flow through system, and we ran water through the tanks each day for one hour in the morning and one hour in the evening to ensure precise control of salinities. At the observed flow rate, this was sufficient for more than one complete exchange daily. Tanks were kept in two water tables to ensure uniform temperatures between tables and replicates. Tanks were moved within and between tables weekly to further reduce the potential for position effects. Lights were on a 12:12 light:dark cycle with gradual transitions to simulate natural conditions. Salinity was measured using a Hach meter Model 60 d. Salinity and temperature were measured daily in a randomly selected 10% of tanks. The mean temperature for all measured tanks was 25.3°C (± 0.03 S.E.). Low salinity tanks were maintained at a mean of 9.4 (± 0.07) ppt. Normal seawater tanks had an average of 29.4 (± 0.50) ppt. Animals were fed ad libidum with Selco-enriched [ 36 ] Artemia (Aquafauna Biomarine, Hawthorne, CA USA).
Experimental design
Phase 1 - population establishment and expansion.
Experimental aquariums were established as matched pairs, one serving as a control (permissive environment) and one subjected to low salinity (stressful environment). Experimental populations were founded with 12 individuals (see above) and these were allowed to breed and expand for three weeks in a permissive environment (~30 ppt salinity).
Phase 2 -- chronic low salinity stress
After this initial census, designated experimental populations were subjected to a stressful environment by gradually reducing the salinity to 9 ppt over the course of four days. Salinity was maintained at this level thereafter. The remaining control tanks were maintained with normal seawater. During the experimental period, a weekly census was conducted in which all individuals were counted and the presence of neonates (animals < 7 days old) was noted.
Fifteen pairs of low diversity (1x) lines were established. We intended to establish these cultures from 15 independently bottlenecked lines, however one of the designated lines went extinct before the start of the experiment, so one of the surviving lines was used twice. Fifteen independent pairs of 2x cultures were also established. Higher diversity levels (6x, 8x and Admixed) were replicated 10 times. The entire experiment contained 120 tanks. A summary of the experimental design is shown in Table 1 .
At the end of the 14-week survey period, surviving individuals were preserved in 100% ethanol from each tank for molecular analysis.
Adaptation over time
To estimate the response to selection of each nominal genetic diversity level over the course of the experiment, population sizes in the stressful and permissive environments were compared three weeks (~1 full reproductive cycle) after the environmental stress was introduced and at the end of the experiment (~3 reproductive cycles later).
Genetic analysis
AFLP genotypes generated from surviving control populations at the end of the experiment were used as a measure of starting genomic diversity for each diversity level. It was not possible to genotype the founding populations at the beginning of the experiment because the low diversity stock lines had only a modest number of individuals, and most of these were required to found the experimental populations. For the lowest diversity lines, the harmonic mean population size was 33.8 individuals, which suggests that the populations would have lost about 2% of their heterozygosity due to genetic drift each generation. In the highest diversity populations, N e was estimated to be 110.6 individuals, consistent with a decline in neutral locus heterozygosity of less than 1% per mysid generation. Some lines were excluded from the molecular analysis because fewer than 10 individuals were available.
Ten individuals were randomly chosen from each line to estimate genetic diversity. DNA was extracted from whole A. bahia using DNeasy ® Blood and Tissue kit (Qiagen, Valencia, CA, USA). The manufacture's instructions were followed except that we heated the elution Buffer AE to 70°C for 10 minutes and incubated the sample with Buffer AE for five minutes at room temperature before eluting each DNA sample. Genomic DNA was quantified using Quant-iT™ PicoGreen ® dsDNA Assay Kit (Invitrogen, Carlsbad, CA, USA) with a Synergy™ HT Multi-Mode Microplate Reader (BioTek, Winooski, VT, USA).
AFLP analysis followed the procedure of Vos et al. [ 35 ], modified to accommodate fluorescent visualization and using the restriction enzyme pair EcoRI/PstI [ 41 ]. Total genomic DNA (75 -- 200 ng) was simultaneously digested and ligated in a 15 μl reaction that included 5 units each of EcoRI, PstI, and T4 DNA ligase (New England Biolabs), 30 pmoles of each EcoRI and PstI double-stranded DNA adaptor [see [ 41 ]], 50 ng/ul BSA, and 50 mM NaCl in T4 Ligase buffer (New England Biolabs). Following complete digestion and ligation at room temperature, products were diluted ten-fold into 10 mM Tris pH 7.6, 0.1 mM EDTA.
Initial PCR enrichment of a subset of fragments (pre-amplification) used 5 μl of the diluted digestion-ligation product as template: 0.5 μM of the EcoRI + A/PstI + C primers (IDT, Coralville, IA) and 0.25 U Taq DNA polymerase (Invitrogen) in 20 μl of 20 mM Tris-HCl (pH 8.4), 50 mM KCl, 0.2 mM each dNTP, and 1.5 mM MgCl 2 . PCR conditions were 2 min at 74°C; 24 cycles of 94°C for 30 sec, 56°C for 30 sec; 72°C for 1 min; followed by 30 sec at 72°C. The pre-amplification product was then diluted ten-fold with 10 mM Tris pH 7.6, 0.1 mM EDTA buffer.
Selective amplification reactions were similar to pre-amplifications, with 3 μl of diluted pre-amplification product used as template and substituting 50 pM of the appropriate FAM--labeled EcoRI + 3/250 pM PstI + 2 selective AFLP primers. Three selective primer combinations were used on all samples: EcoRI + ACT--PstI + CT; EcoRI + AGG--PstI + CA; and EcoRI + ATG--PstI + CT. PCR conditions were 2 min at 94°C, 12 cycles of 20 sec at 94°C, 30 sec at 66°C dropping 1°C per cycle, 1 min at 72°C; then 20 cycles of 20 sec at 94°C, 30 sec at 56°C, 1 min at 72°C; followed by 30 min at 72°C. AFLP genotypes were electrophoresed and visualized with an ABI 3730 DNA analyzer.
Bins within the range of 100 to 500 bp [ 38 ] were generated for the amplified fragments using GeneMarker ® version 1.6 (SoftGenetics LLC ® , State College, PA, USA). We manually checked the quality of each AFLP fingerprint and bin using the method described by Whitlock et al. [ 39 ] with slight modifications. We removed samples that produced an AFLP fingerprint with less than 20 peaks within the target size range and restricted our analyses to fragments with relative florescence units greater than 100 to reduce background noise. We visually checked the automatically created bins to ensure the bin was centered on the distribution of peaks within the bin and removed bins that had AFLP fragments that differed in size by more than 1 bp. We also deleted bins with fragment-length distributions that overlapped with adjacent bins to reduce the occurrence of homoplasy [ 38 , 40 ]. The number of initial bins for the three sets of restriction enzymes ranged from 63 to 76 each. We developed an R http://www.R-project.org/ script to convert the raw peak intensity data output from GeneMarker to a format compatible for AFLPScore version 1.3 [ 40 ]. We scored our raw AFLP data using AFLPScore, normalized our data to the median, filtered our data with a locus selection threshold, and used a relative genotype calling threshold. We tested a range of locus (100 to 1000 bp) and genotype thresholds (1 to 120%) and selected the pair of values that simultaneously minimized the mismatch error rate, minimized the probability of misscoring a presence allele (ε 1.0 error rate), and maximized the number of loci retained. We included all pairwise comparisons for the samples that had greater than two replicates in our mismatch analysis. We generated AFLP genotypes for each restriction enzyme pair with the optimized locus selection and genotype thresholds using AFLPScore.
The locus selection threshold was 1000 bp and the genotype threshold was 10% for each restriction enzyme pair. The average mismatch error rate for the three restriction enzyme pairs was 8.3504 ± 1.7367 (S.D.) and the average ε 1.0 error rate was 19.484 ± 2.3992. Following the intensive screening and quality control process, 59 loci (bins) were available to estimate genetic diversity. AFLP based estimates of genetic diversity were calculated using AFLP-Surv v1.0 [ 38 ]. AFLP based estimates of genetic diversity were calculated as either the fraction of polymorphic loci within the sample (PLP) or the heterozygosity analogue (H j ) [ 41 ].
Statistical analyses
Three different indices of population fitness were evaluated: 1) the number of individuals in the Last Census (LC), 2) the Median Population Size (MPS) using data from all 13 censuses for each experimental tank and 3) the Reproductive Index (RI), which was calculated as the number of weeks in which reproduction was observed divided by the total number of weeks that the population survived for each population.
Statistical relationships among fitness, genetic diversity (treating levels 1x, 2x, 6x, 8x, and Admixed as ordinal categorical data), and environmental stress were evaluated using general linear models. All calculations were performed using either JMP 7.0 or SAS 8.0 (SAS institute, Cary NC).
The genetic load of the inbred 1x was estimated relative to the outcrossed AMX lines using the methods of Morton and Crow [ 42 ]. Genetic load was not estimated during the creation of the inbred lines because reference populations required for the calculation were not established due to space limitations.
Genetic Load and Extinction of Founder Lines
A substantial proportion of the bottlenecked lines either failed to thrive or did not survive long enough to be used in the main experiment. Of the 64 lines initiated, only 14 achieved a population size of at least 26 individuals, the predetermined threshold deemed sufficient to generate dihybrid lines and maintain the 1x lines.
Genetic load within the main experiment was estimated to be higher in the stressful environment for all three fitness indices. Using LC, the number of lethal equivalent loci in the permissive environment was 2.87 compared to 10.97 in the stressful environment. Lethal equivalents for MPS were 3.45 in the permissive environment and 6.71 in the stressful environment. For RI, in a permissive environment we estimate that there were 1.06 lethal equivalents compared to 3.19 in the stressful environment.
Molecular estimates of genetic diversity
After screening, a total of 59 AFLP markers were available for analysis. Average PLP for the 1x lines was 35.6 ± 7.3 (S.D.) and average H j was 0.14 ± 0.05. In comparison, AMX lines had an average PLP of 52.1 ± 4.3 and an average H j of 0.19 ± 0.02. Nominal genetic diversity explained a moderate amount of variation in AFLP diversity estimates (PLP Spearman's ρ = 0.67, p < 0.0001; H j Spearman's ρ = 0.44, p = 0.0043). In post-hoc tests, neither estimator was effective at differentiating among the three highest genetic diversity treatments; however the 1x, 2x and higher diversity lines were distinguishable from each other when PLP was used to estimate genetic diversity (Table 2 ).
Population growth in permissive conditions
Abundance after three weeks of culture under permissive conditions (Phase 1) was significantly correlated with nominal genetic diversity level (Spearman's ρ = 0.68, p < 0.0001, Table 3 ). Final population sizes increased from 12 individuals at the start of the experiment to an average of 18.6 individuals in the low diversity lines (1x) and to 79.3 individuals in the highest diversity populations (AMX). All treatments differed from each other, except 6x and 8x. Variance was unequal among treatments (p = 0.0244) with the coefficient of variation inversely related to genetic diversity (Table 3 , Figure 1 ).

Population fitness, estimated with Median Population Size (A), Last Census size (B), and Reproductive Index (C) . Paired box plots define the median and middle two quantiles in stressful (left) and permissive environments (right). Lower case letters unite groups that are not statistically distinguishable using post-hoc tests (Tukey's HSD) at α = 0.05.
AFLP diversity estimated as PLP explained a modest percentage of the variation in abundance after three weeks in permissive conditions (adjusted R 2 = 0.24, p < 0.0001). AFLP diversity estimated as H j explained less but still significant abundance variation (adjusted R 2 = 0.16, p < 0.0001).
Population fitness, environmental stress and genetic diversity
A model including genetic diversity and environmental stress explained much of variation in MPS during the chronic low salinity experiment (Phase 2) (adjusted R 2 = 0.74, p < 0.0001). Both factors contributed strongly to the relationship (environment F = 127.8, p < 0.0001; diversity F = 53.3, p < 0.0001). Treatment means ranged from 9.7 individuals (low salinity, 1X) to 123.2 individuals (normal salinity, AMX). There was no significant interaction between salinity stress and genetic diversity level (F = 0.59, p = 0.6693) (Figure 1 ).
An additional model that included the results of the first census (Phase 1, pre-stress) as a covariate also explained much of the variation in MPS (Adj R 2 = 0.78, p < 0.0001). Abundance at initiation of experimental treatments was a significant covariate (F = 16.5, p < 0.0001). In this more complex model, there was a significant interaction between initial abundance and genetic diversity level (F = 4.0, p = 0.0043), but no interaction between environment and genetic diversity (F = 0.78, p = 0.536). Both nominal diversity level (F = 13.1, p < 0.0001) and environment (F = 22.3, p < 0.0001) were significant individually.
The last census sizes ranged from a mean of 2.5 individuals (low salinity, 1x) to 84.4 individuals (normal salinity, AMX). A model including nominal genetic diversity and environmental stress explained 53% of the observed variation in LC (p < 0.0001). Genetic diversity (F = 50.8, p < 0.001) and environmental stress (F = 21.4, p < 0.001) were both significant, but the interaction term was not (F = 0.54, p = 0.71). An expanded model for LC that included the results of the first census (Phase 1, pre-stress) as a covariate explained no additional variation in LC (Adj R 2 = 0.53, p < 0.0001), and the covariate was marginally insignificant (F = 3.45, p < 0.0658).
Environmental stress and genetic diversity explained much of the variation in RI (adjusted R 2 = 0.58, p < 0.0001). Both factors were statistically significant (stress F = 95.4, p < 0.0001; genetic diversity F = 14.3, p < 0.0001) with a marginally insignificant interaction between these two factors (F = 2.18, p = 0.0751).
In these analyses, variance was unequal among treatments, and remained unequal despite attempted transformations. Variance was higher among low diversity populations and under stressed conditions (Figure 2 ). Variation (expressed as the coefficient of variation) in all three fitness proxies is summarized in Table 3 , and the distribution of individual replicate values is shown in Figure 1 . The most likely effect of unequal variances in these analysis is an increase in Type I error, which could be compensated for by reducing α by half to 0.025 [ 43 ]. All effects that were previously found to be significant remain significant under this more stringent criterion.

Ratios of census sizes in the stressful environment to those in the permissive environments for each diversity class after three weeks in the selective environment (left box plot) and at the end of the experiment (right box plot) . The box plots enclose the central two quantiles and show the group medians. Only the 1x and 2x treatments are significantly distinguishable using the Wilcoxon signed rank test. The inset shows the average percent decline in census size in the stressed populations relative to the control populations at the end of the experiment.
Population fitness, environmental stress and molecular diversity
The effects of AFLP diversity (estimated for each individual replicate using either PLP or H j ) and environmental stress were evaluated for three different fitness indices: MPS, LC, and RI.
A significant portion of the variation in MPS is explained by a model incorporating AFLP diversity measured as PLP and environmental stress (adjusted R 2 of 0.53, p < 0.0001). Both variables were statistically significant (stress F = 59.1, p < 0.0001; PLP F = 32.9, p < 0.0001), and there was no significant interaction between the two terms (F = 0.04, p = 0.83). Similar results were obtained when H j was substituted for PLP (adjusted R 2 = 0.51, p < 0.001; stress F = 56.1, p < 0.0001; H j F = 27.2, p < 0.0001; stress*H j F = 0.26, p = 0.61).
Models evaluating the effect of AFLP diversity and environmental stress on LC were also significant overall (adjusted R 2 = 0.37, p < 0.0001 using PLP, adjusted R 2 = 0.38, p < 0.0001 using H j ). There was no interaction between genetic diversity and stress in either model using PLP (PLP F = 11.8, p < 0.0001; stress F = 36.8, p = 0.001; stress*PLP F = 0.58, p = 0.45) or H j (H j F = 13.5, p < 0.0004; stress F = 37.2, p < 0.0001; stress*H j F = 0.0009, p = 0.97).
Similarly, both PLP and H j explained a significant fraction of the variation in RI (PLP Adj R 2 = 0.53, environment F = 75.6, p < 0.0001, PLP F = 15.6, p = 0.0002), (H j Adj R 2 = 0.52, environment F = 73.9 p < 0.0001, H j F = 14.0, p = 0.0002). Neither genetic diversity estimator had a significant interaction with environmental stress.
Observed population extinctions
Population extinctions were rare during the course of the study, and were confined to the low diversity populations (Table 3 ). Three out of 15 1x populations went extinct under permissive conditions. Median time to extinction for these populations was seven weeks. By contrast, 11 of 15 1x populations went extinct under stressful conditions, with a median extinction time of nine weeks. Only a single 2x population went extinct in the low salinity treatment at 11 weeks.
AFLP data were available for nine of the 15 pairs in the lowest diversity 1x treatment. The remaining six pairs could not be surveyed due to extinction or low survivor numbers in the control line. The lines that went extinct had a mean PLP of 32.5 compared to 39.4 for the surviving lines, although this difference was not significant (p = 0.17). H j in extinct lines was 0.11 and 0.18 in surviving lines, and the difference was statistically significant (p = 0.014) (Table 2 ).
After three weeks exposure to low salinity (Week 6 of the experiment), the average census size for 1x populations in this stressful environment was 57% smaller than those in the high salinity control environment, while the high diversity AMX lines were, on average, 24% smaller in the stressful environment relative to their controls. At the end of the experiment (Week 16), census sizes for 1x stressed populations were 94% smaller than their controls on average, whereas average AMX populations reared in low salinity was only 7% smaller than their controls. The relative decline in census size of the salinity stressed 1x populations was partly driven by the extinct lines; however, when these were excluded the net decline relative to the control population was still 83% (Figure 2 ).
The experimental results presented here indicate that the Americamysis bahia system for generating defined levels of genetic diversity with a high level of replication is a useful tool for addressing empirical questions in conservation genetics. The results from this initial experiment measure: 1) the relative performance of high and low diversity populations in both good and bad environments; 2) the power of AFLP markers to detect meaningful losses of genetic diversity; 3) the magnitude of genetic load in both good and bad environments; 4) the potential utility of genetic rescue and heterosis; and 5) the relative potential for adaptation to novel environments.
Reduced diversity and population fitness
In this simplified laboratory environment, lower population genetic diversity was associated with lower population fitness, although this decrease was not always statistically significant in all post-hoc tests. As expected, average population fitness in the stressful environment was always lower than fitness in the permissive environment for a given level of genetic diversity.
Interestingly, none of the interaction terms between nominal genetic diversity and environmental stress were significant for any of our three fitness indices although the interaction was significant for RI. This may indicate that factors interact less in our laboratory setting than might be expected in a more complex natural environment, but we cannot rule out the possibility of insufficient statistical power. Power analysis could potentially address this issue, however we lack a non-arbitrary estimate of the magnitude of a meaningful effect [ 44 ]. Similarly, we did not detect an interaction between either of the AFLP based diversity estimates and the environment when estimating fitness. This particular analysis is complicated by the fact that extinctions in some of the 1x control lines reduced the number of lines available with very low diversity.
A modest amount of neutral locus genetic diversity (as estimated with AFLP genotypes) was explained by nominal diversity level. The overall relationship was in the expected direction; however, post hoc tests (Table 2 ) reveal that estimates of both PLP and H j based on our final set of 59 screened AFLP markers do not reliably detect differences between the three highest nominal diversity levels. Similarly, both estimators explain only a modest amount of the variation in the three fitness indices. Despite the lack of a detectable molecular genetic difference, the observed mean fitness was always lower in 8x populations than in AMX populations in the stressful environment and for two of the three fitness proxies in the permissive environment. Post-hoc tests showed these differences were statistically significant for two of the three proxies in the stressful environment (Figure 1 ).
In this simplified experimental environment, AFLP markers detected large decreases in genetic diversity but missed more modest but ecologically meaningful losses. This may have important implications for the application of AFLP genotypes. While AFLP markers lack the power to detect all meaningful losses of genetic diversity, these markers are unlikely to cause false positives; when detectable losses in AFLP diversity occur, our data suggest they signal a serious decline in population viability.
Inbreeding, Genetic load and Hybrid Rescue
The clearest evidence for the effects of inbreeding on A. bahia populations was obtained before the formal experiment started. In order to generate the 1x lines used in this study, we started with 64 founding lines. Fully three quarters of these lines failed to generate the 26 individuals that were required to found the experimental lines after several months in culture. Some early losses may also be due to demographic stochasticity--initial brood sizes are small in young mysid females. However many lines that survived failed to thrive during more than four months under permissive conditions. Thus, inbreeding effects were a major determinant of the number and types of lines available for our main experiment making it necessary to construct experimental populations using only the modest number of lines that were most resistant to inbreeding depression. This result is typical of animals with large, panmictic populations [ 24 , 45 ].
From a conservation genetics perspective, it is important to understand the population level consequences of individual inbreeding depression (or the approximate opposite, heterosis). It has been repeatedly noted that the impact of individual inbreeding depression varies with environment [ 46 , 47 ], and the negative effects of high levels of inbreeding may be masked by permissive environments or when a direct comparison with outbred individuals is not possible [ 6 , 12 , 48 ]. In experimental settings, inbreeding depression is usually, but not universally, stronger in stressful environments [ 46 ]. In the mysid experimental system, estimates of the genetic load in both environments suggest that while inbreeding depression is expressed for all fitness metrics in the permissive environment, the effects are far more pronounced in the stressful environment. We note that owing to experimental constraints, this estimate of genetic load is applicable only to the main experiment and does not necessarily reflect genetic load within natural populations.
Because we constructed our higher diversity populations by combining different numbers of low diversity lines, our study may be viewed as a series of replicated 'genetic rescue'[ 49 ] experiments (albeit with very high immigration rates, comparable to [ 45 ]). Population fitness was substantially improved when two or more 1x lines were combined, and in almost all cases, the 'rescue' was successful. Only a single 2x population went extinct in the stressful environment. Within our system, nominal genetic diversity was an important predictor of population fitness for most levels of genetic diversity. In both environments and for all three of the fitness proxies, the 2x lines performed better on average than the 1x lines, and the 6x lines performed better than the 2x lines. The difference was not always statistically significant in post-hoc comparisons for each fitness index at each level (Figure 1 ), but the relative performance was as expected. Further, the high diversity AMX populations were generally more fit than any of the lower diversity populations.
We did not detect a statistically significant difference between the 6x and 8x populations in any of the fitness assays or by using molecular markers. We note that the best performing 8x were superior to the best performing 6x populations, however the worst performing 8x populations were inferior to the worst performing 6x populations. Because the 8X lines were founded with only three individuals from each of four founding 2x lines, it is possible that some of the founding lines did not establish themselves in some 8x populations. In any case, genetic diversity levels in these two classes are expected to be quite similar. Even for a locus that is fixed for alternate alleles in the 1x populations, expected heterozygosity of 6x and 8x populations would only differ by 4% on average (H = 0.833 and 0.875, respectively [ 50 ]). The actual heterozygosity difference is likely under 2% since 1x lines would have experienced only a 30% to 50% reduction in heterozygosity relative to the founding stock populations.
Diversity, selection and adaptation
Many studies have focused on the individual fitness consequences of inbreeding in benign and stressful environments due to inbreeding depression effects [ 46 ] but this is only one way that genetic diversity affects extinction risk. It also is important to determine the consequences of reduced genetic diversity for the capacity of the population to adapt to a novel environment. Even modest losses of genetic diversity may result in a reduced ability to adapt to environmental change, yet the short-term impact of such losses may be minimal if populations are maintained in stable environments or if the loss does not cause detectable inbreeding depression-like effects. The long-term impact of moderate losses on population persistence can best be measured by estimating generational changes in population fitness in multiple environments. The mysid experimental system demonstrates that both population fitness and inter-population variability are influenced by genetic diversity, and that both fitness and variability are influenced by environmental stress.
To assess the strength of selection in the stressful environment, we calculated the ratio of populations in the stressful environment to those in the permissive environment three weeks (~1 mysid generation) after the stressful environment was introduced. We hypothesized that the relative proportions should be similar at both time points if inbreeding and heterosis are influencing the relationship, but that when adaptation has occurred, population sizes in the stressful and permissive environments will grow more similar over time. We found that after three weeks of selection the 1x population sizes in the stressful environment were 57% smaller than those in the permissive environment, while the AMX population sizes were only 24% smaller in the stressful environment. These declines represent the selection pressure imposed by the stressful environment. After 10 more weeks of selection, the AMX population sizes in the stressful environment were only 7% lower than those in the permissive environment while the 1x population sizes were 94% lower (Figure 2 ). Therefore, the low diversity populations did poorly in the stressful environment early in the experiment and grew progressively worse as the experiment proceeded. By contrast, the high diversity populations were relatively less disadvantaged early on and even showed some improvement by the end of the experiment. In the AMX lines the level of improvement did not rise to statistical significance; however, the trend was consistent with the one predicted by evolutionary adaptation (and some stressed populations even outperformed their matched controls), suggesting that simple heterosis may not be the only force operating in populations with high genetic diversity. However, these results should be interpreted with some caution as the high diversity populations may have been close to the carrying capacity of the habitat in both normal and low salinity environments.
In our mysid data set, nominal genetic diversity was an important predictor of variability between populations within an environmental treatment with lower diversity populations having more inter-population variability than higher diversity populations. Population size (either median or final) was also notably lower in low genetic diversity populations, so a much higher fraction of low diversity lines are likely to fall below the minimum number of individuals required to maintain population viability [ 51 ]. In general, temporal variation in abundance within a single population is expected to increase the chances of population loss [ 51 , 52 ], so these results indicate that genetic diversity is an important component of extinction risk.
Using the mysid experimental system, we found that: 1) reduced population genetic diversity reduces population fitness in both permissive and stressful environments; 2) even some modest reductions in genetic diversity can reduce the value of some fitness measures, especially in stressful environments; 3) environmental stress and genetic diversity appear to independently influence population fitness; and 4) AFLP genotypes detected large reductions in population genetic diversity, but did not reliably detect modest reductions in genetic diversity that may influence population fitness. Therefore, many more AFLP loci than are commonly used would be necessary to detect these losses. However when genetic diversity losses are detected using a moderate number of AFLP loci, they are likely to be ecologically important. We also found that: 5) low diversity populations show more inter-population variability than high diversity populations for most estimates of population fitness; and 6) high diversity populations showed some capacity to adapt to the stressful environment, but low diversity populations did not.
In natural populations the relationship between population fitness and genetic diversity will depend on specifics of the environment and the organism. Genetic diversity may not always enable populations to persist, but a lack of diversity essentially guarantees that adaptation to altered environments will not occur. Despite the importance of diversity for population survival, our understanding of the relationship between diversity and long-term population viability is limited. Studies in simplified laboratory environments, such as the one described here, can be used to determine a baseline for the relationship between diversity and population risk under the best possible conditions (i.e., with the least environmental variation) and provide an important way to assess molecular tools that are potentially useful in conservation biology.
Abbreviations
Admixed lines
Heterozygosity estimate derived from dominant molecular markers
The Last Census; the number of individuals in an aquarium at the end of the experiment
Median Population Size for a single line over the course of the experiment
Net Increase in population size after three weeks in permissive conditions
Proportion of Loci Polymorphic; the fraction of AFLP bands that vary within an experimental populations
Reproductive Index; the fraction of census weeks in which neonates were observed
Time To Extinction.
Maehr DS, Crowley P, CJ J, LM J, Larkin JL, Hoctor TS, Harris LD, HP M: Of cats and haruspices: genetic intervention in the Florida panther. Response to Pimm et al . (2006). Animal Conservation. 2006, 9: 127-132. 10.1111/j.1469-1795.2005.00019.x.
Article Google Scholar
Westemeier R, Brawn J, Simpson S, Esker T, Jansen R, Walk J, Kershner E, Bouzat J, Paige K: Tracking the long-term decline and recovery of an isolated population. Science. 1998, 282: 1695-1698. 10.1126/science.282.5394.1695.
Article CAS PubMed Google Scholar
Gilpin ME, Soule ME: Minimum viable populations: processes of species extinction. Conservation biology: the science of scarcity and diversity. Edited by: Soule ME. 1986, Sunderland, MA: Sinauer Associates Inc
Google Scholar
Ellstrand NC, Elam DR: Population size: Implications for plant conservation. Annual Review of Ecology and Systematics. 1993, 24: 217-242. 10.1146/annurev.es.24.110193.001245.
Boakes EH, Wang J, Amos W: An investigation of inbreeding depression and purging in captive pedigreed populations. Heredity. 2007, 98: 172-182. 10.1038/sj.hdy.6800923.
Fritzsche P, Neumann K, Nasdal K, Gattermann R: Differences in reproductive success between laboratory and wild-derived golden hamsters (Mesocricetus auratus) as a consequence of inbreeding. Behavioral Ecology and Sociobiology. 2006, 60: 220-226. 10.1007/s00265-006-0159-3.
Johnson JA, Dunn PO: Low genetic variation in the heath hen prior to extinction and implications for the conservation of prairie-chicken populations. Conservation Genetics. 2006, 7: 37-48. 10.1007/s10592-005-7856-8.
Pray LA, Schwartz JM, Goodnight CJ, Stevens L: Environmental dependency of inbreeding depression: implications for conservation biology. Conservation Biology. 1993, 8: 562-568. 10.1046/j.1523-1739.1994.08020562.x.
Slate J, Kruuk LEB, Marshall TC, Pemberton JM, Clutton-Brock TH: Inbreeding depression influences lifetime breeding success in a wild population of red deer ( Cervus elaphus ). Proceedings of the Royal Society of London, Series B. 2000, 267: 1657-1662. 10.1098/rspb.2000.1192.
Article CAS Google Scholar
Bijlsma R, Bundgaard J, Boerema AC, Van Putten WF: Genetic and environmental stress, and the persistence of populations. Environmental Stress, Adapation and Evolution. Edited by: Loeschcke RBaV. 1998, Basel: Birkhauser Verlag
Reed DH, Briscoe DA, Frankham R: Inbreeding and extinction: the effect of environmental stress and lineage. Conservation Genetics. 2002, 3: 301-307. 10.1023/A:1019948130263.
Keller LF, Grant PR, Grant BR, Petren K: Environmental conditions affect the magnitude of inbreeding depression in survival of Darwin's finches. Evolution. 2002, 56: 1229-1239.
Article PubMed Google Scholar
Da Silva A, Luikart G, Yoccoz NG, Cohas A, Allaine D: Genetic diversity-fitness correlation revealed by microsatellite analyses in European alpine marmots (Marmota marmota). Conservation Genetics. 2006, 7: 371-382. 10.1007/s10592-005-9048-y.
Lippman ZB, Zamir D: Heterosis: revisiting the magic. Trends in Genetics. 2007, 23: 60-66. 10.1016/j.tig.2006.12.006.
Markert JA, Grant PR, Grant BR, Keller LF, Coombs JL, Petren K: Neutral locus heterozygosity, inbreeding, and survival in Darwin's ground finches (Geospiza fortis and G. scandens). Heredity. 2004, 92: 306-15. 10.1038/sj.hdy.6800409.
Slate J, David P, Dodds KG, Veenvliet BA, Glass BC, Broad TE, McEwan JC: Understanding the relationship between the inbreeding coefficient and multilocus heterozygosity: theoretical expecations and empirical data. Heredity. 2004, 93: 255-265. 10.1038/sj.hdy.6800485.
Nacci D, Champlin D, Coiro L, McKinney R, Jayaraman S: Predicting the occurrence of genetic adaptation to dioxinlike compounds in populations of the estuarine fish Fundulus heteroclitus . Environmental Toxicology and Chemistry. 2002, 21: 1525-1532.
Fischer JM, Klug JL, Ives AR, Frost TM: Ecological history affects zooplankton community responses to acidification. Ecology. 2001, 82: 2984-3000. 10.1890/0012-9658(2001)082[2984:EHAZCR]2.0.CO;2.
Hoffmann AA, Hallas RJ, Dean JA, Schiffer M: Low potential for climatic stress adapation in a rainforest Drosophila species. Science. 2003, 301: 100-102. 10.1126/science.1084296.
Gienapp P, Postma E, Visser ME: Why breeding time has not responded to selection for earlier breeding in a songbird population. Evolution. 2006, 60: 2381-2388.
Leberg P: Influence of genetic variability on population growth: implications for conservation. Journal of Fish Biology. 1990, 37 (Supplement A): 193-195. 10.1111/j.1095-8649.1990.tb05036.x.
Leberg P: Strategies for population reintroduction: Effects of genetic variability on population growth and size. Conservation Biology. 1993, 7: 194-199. 10.1046/j.1523-1739.1993.07010194.x.
Newman D, Pilson D: Increased probability of extinction due to decreased genetic effective population size: Experimental populations of Clarkia pulchella . Evolution. 1997, 51: 354-362. 10.2307/2411107.
Bryant EH, Backus VL, Clark ME, Reed DH: Experimental tests of captive breeding for endangered species. Conservation Biology. 1999, 13: 1487-1496. 10.1046/j.1523-1739.1999.98440.x.
Frankham R, Lees K, Montgomery ME, England PR, Lowe EH, Briscoe DA: Do population size bottlenecks reduce evolutionary potential. Animal Conservation. 1999, 2: 10.1111/j.1469-1795.1999.tb00071.x.
Frankham R: Stress and adaptation in conservation genetics. Journal of Evolutionary Biology. 2005, 18: 750-755. 10.1111/j.1420-9101.2005.00885.x.
Ludovic A, Brostaux Y, Lallemand S, Haubruge E: Reproductive strategies of Tribolium flour beetles. Journal of Insect Science. 2005, 5: 33-
Tatar M, Promislow DEL, Khazaeli AA, Curtsinger JW: Age-specific patterns of genetic variance in Drosophila melanogaster . II. Fecundity and its genetic covariance with age-specific mortality. Genetics. 1996, 143: 849-858.
PubMed Central CAS PubMed Google Scholar
Heard RW, Knott DM, King RA, Allen DM: A taxonomic guide to the mysids of the South Atlantic. Book A taxonomic guide to the mysids of the South Atlantic. (Editor ed.^eds.). 2006, City: U.S. Department of Commerce, 37-
McKenney CL: Optimization of environmental factors during the life cycle of Mysidopsis bahia . EPA Environmental Research Brief. 1987, EPA/600/M-87/004: 1-6.
McKenney CL: The combined effects of salinity and temperature on various aspects of the reproductive biology of the estuarine mysid, Mysidopsis bahia . Invertebrate reproduction and development. 1996, 29: 9-18.
Kuhn A, Munns WR, Champlin D, McKinney R, Tagliabue M, Serbst J, G T: Evaluation of the efficacy of extrapolation population modeling to predict the dynamics of Americamysis bahia populations in the laboratory. Environmental Toxicology and Chemistry. 2001, 20: 213-221.
Lussier SM, Kuhn A, Chammas MJ, Swewall J: Techniques for the laboratory culture of Mysidopsis species (Crustacea: Mysidae). Environmental Toxicology and Chemistry. 1988, 7: 969-977.
Odum WE, Heald EJ: Trophic analysis of an estuarine mangrove community. Bulletin of Marine Science. 1972, 22: 671-738.
Vos P, Hogers R, Bleeker M, reijans M, Lee TVd, Hernes M, Frijters A, Pot J, Peleman J, Kuiper M: AFLP: a new technique for DNA fingerprinting. Nucleic Acids Research. 1995, 23: 4407-4414. 10.1093/nar/23.21.4407.
Article PubMed Central CAS PubMed Google Scholar
Kuhn AH, Bengtson DA, Simpson KL: Increased reproduction by Mysids ( Mysidopsis bahia ) fed with enriched Artemia spp. Nauplii. American Fisheries Society Symposium. 1991, 9: 192-199.
Frankham R, Ballou JD, Briscoe DA: Introduction to conservation genetics. 2004, Cambridge: Cambridge University Press
Vekemans X, Beauwens T, Lemaire M, Roldan-Ruiz I: Data from amplified fragment length polymorphism (AFLP) markers show indication of size homoplasy and of a relationship between degree of homoplasy and fragment size. Molecular Ecology. 2002, 11: 131-151. 10.1046/j.0962-1083.2001.01415.x.
Whitlock MC: Nonequilibrium population structure in forked fungus beetles: extinction, colonization and the genetic variance among populations. American Naturalist. 1992, 139: 952-970. 10.1086/285368.
Whitlock R, Hipperson H, Mannarelli M, Butlin RK, Burke T: An objective, rapid and reproducible method for scoring AFLP peak-height data that minimizes genotyping error. Molecular Ecology Resources. 2008, 8: 725-735. 10.1111/j.1755-0998.2007.02073.x.
Lynch M, Milligan BG: Analysis of populationgenetic structure with RAPD markers. Molecular Ecology. 1994, 3: 91-99. 10.1111/j.1365-294X.1994.tb00109.x.
Morton NE, Crow JF, Muller HJ: An estimate of the mutational damage in man from data on consanguineous marriages. Proceedings of the National Academy of Science. 1956, 42: 855-863. 10.1073/pnas.42.11.855.
Keppel G, Wickens TD: Design and Analysis. 2004, Upper Saddle River, NJ, Prentice Hall, 4
Goodman SN, Berlin JA: The use of predicted confidence intervals when planning experiments and the misuse of power when interpreting results. Annals of Internal Medicine. 1994, 121: 200-206.
Backus VL, Bryant EH, Hughes CR, Meffert LM: Effect of migration or inbreeding followed by selection on low-founder-number populations: implications for captive breeding programs. Conservation Biology. 1995, 9: 1216-1224. 10.1046/j.1523-1739.1995.9051216.x.
Armbruster P, Reed D: Inbreeding depression in benign and stressful environments. Heredity. 2005, 95: 235-242. 10.1038/sj.hdy.6800721.
Cheptou PO: The ecology of inbreeding depression. Heredity. 2006, 96: 110-10.1038/sj.hdy.6800768.
Keller LF, Waller DM: Inbreeding effects in wild populations. Trends in Ecology and Evolution. 2002, 17: 230-241. 10.1016/S0169-5347(02)02489-8.
Tallmon DA, Luikart G, Waples RS: The alluring simplicity and complex reality of genetic rescue. Trends in Ecology & Evolution. 2004, 19: 489-496.
Pederson DG: The expected degree of heterozygosity in a double-cross hybrid population. Genetics. 1966, 53: 669-674.
Lande R, Engen S, B-E Saether: Stochastic Population Dynamics in Ecology and Conservation. 2003, Oxford: Oxford University Press
Book Google Scholar
Lewontin RC, Cohen D: On population growth in a randomly varying evironment. Proceedings of the National Academy of Science. 1969, 69: 1056-1060. 10.1073/pnas.62.4.1056.
Download references
Acknowledgements
We appreciate the helpful advice from reviewers of early drafts, including Dina Proestou (U.S. Environmental Protection Agency), John Darling (U.S. Environmental Protection Agency), and Rebecca Brown (University of Rhode Island). Animal collection was supported by welcome advice and assistance from Sandy Raimondo (U.S. Environmental Protection Agency). Suzy Ayvazian, Danielle Duquette, Peg Pelletier, Mark Tagliabue, Glen Thursby, Jon Serbst, and Sarah Winnicki all helped count or culture animals while Suzanne Jackson helped support the AFLP genotyping efforts. This is contribution number AED-07-102 of the U.S. Environmental Protection Agency, Office of Research and Development, National Health and Environmental Effects Research Laboratory, Atlantic Ecology Division, which partially supported this research. This manuscript has been reviewed and approved for publication by the U.S. EPA. Approval does not signify that the contents necessarily reflect the views and policies of the U.S. EPA. Mention of trade names, products, or services does not convey, and should not be interpreted as conveying, official U.S. EPA approval, endorsement, or recommendation.
Author information
Authors and affiliations.
Population Ecology Branch, Atlantic Ecology Division, U.S. Environmental Protection Agency, 27 Tarzwell Dr., Narragansett, RI, USA
Jeffrey A Markert, Denise M Champlin, Ruth Gutjahr-Gobell, Jason S Grear, Anne Kuhn, Thomas J McGreevy Jr & Diane E Nacci
Molecular Ecology Research Branch, Ecological Exposure Research Division, U.S. Environmental Protection Agency, 26 Martin Luther King Dr., Cincinnati, OH, 45268, USA
Jeffrey A Markert, Annette Roth & Mark J Bagley
Department of Natural Resources Science, Coastal Institute, University of Rhode Island, 1 Greenhouse Rd., Kingston, RI, 02881, USA
Thomas J McGreevy Jr
c/o U.S. Geological Survey, San Diego Field Station, Western Ecology Research Center, 4165 Spruance Rd., San Diego, CA, 92101, USA
Jeffrey A Markert
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Jeffrey A Markert .
Additional information
Authors' contributions.
JAM, DEN and MJB took the lead in designing overall experiment, while AK, JSG and DMC provided critical insights into key sections of the design. DMC, AK and JAM also conducted an extensive series of pilot studies that made this project possible. RG-G designed methods that allowed us to precisely maintain the experimental environment. TJM-optimized objective AFLP scoring parameters, produced AFLP genetic diversity estimates, and wrote the AFLP scoring section. AR managed the collection of molecular data and wrote the molecular section of the AFLP methods. All Narragansett based authors participated in weekly censuses and daily culture activities. All authors contributed to the writing of the paper and have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Authors’ original file for figure 1
Authors’ original file for figure 2, rights and permissions.
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License ( http://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Reprints and permissions
About this article
Cite this article.
Markert, J.A., Champlin, D.M., Gutjahr-Gobell, R. et al. Population genetic diversity and fitness in multiple environments. BMC Evol Biol 10 , 205 (2010). https://doi.org/10.1186/1471-2148-10-205
Download citation
Received : 21 September 2009
Accepted : 07 July 2010
Published : 07 July 2010
DOI : https://doi.org/10.1186/1471-2148-10-205
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- AFLP Marker
- Genetic Load
- Population Genetic Diversity
- Permissive Environment
- Population Fitness
BMC Ecology and Evolution
ISSN: 2730-7182
- General enquiries: [email protected]

Genetic structure of restored Brook Trout populations in the Southern Appalachian Mountains indicates successful reintroductions
- Rebecca J. Smith
- David C. Kazyak
- Benjamin M. Fitzpatrick

Correction: Revisiting conservation units for the endangered mountain yellow-legged frog species complex (Rana muscosa, Rana sierrae) using multiple genomic methods
- Allison Q. Byrne
- Andrew P. Rothstein
- Erica Bree Rosenblum
Genomic patterns of native palms from the Leeward Antilles confirm single-island endemism and guide conservation priorities
- James A. R. Clugston
- Quirijn Coolen
- M. Patrick Griffith

Isolation, small population size, and management influence inbreeding and reduced genetic variation in K’gari dingoes
- Susan M. Miller
- Linda Behrendorff
- Jacqueline M. Bishop

Loss of genetic diversity and isolation by distance and by environment in populations of a keystone ungulate species
- Fernanda de Góes Maciel
- Sean O’Rourke
- Cibele Biondo

Early detection of rare and elusive endangered species using environmental DNA: a case study for the Eurasian otter and the white-clawed crayfish in northwestern Italy
- Lorenzo Ballini
- Dario Ottonello
- Alessio Iannucci

Genetic connectivity in the Arizona toad ( Anaxyrus microscaphus ): implications for conservation of a stream dwelling amphibian in the arid Southwestern United States
- Sara J. Oyler-McCance
- Mason J. Ryan

Absence of genetic structure among ecologically diverse populations indicate high plasticity in a pantropical seabird
- Mariana Scain Mazzochi
- Vitória Muraro
- Leandro Bugoni

Genomic and common garden data reveal significant genetic differentiation in the endangered San Fernando Valley spineflower Chorizanthe parryi var. fernandina
- Deborah L. Rogers
- Loraine Kohorn Washburn
- Andrea D. Schreier

Admixture and reproductive skew shape the conservation value of ex situ populations of the Critically Endangered eastern black rhino
- Franziska Elsner-Gearing
- Petra Kretzschmar
- Catherine Walton

Genetic structure and diversity of semi-captive populations: the anomalous case of the Asian elephant
- Gilles Maurer
- Marie-Pierre Dubois
- Finn Kjellberg

eDNA metabarcoding reveals a rich but threatened and declining elasmobranch community in West Africa’s largest marine protected area, the Banc d’Arguin
- Carolina de la Hoz Schilling
- Rima W. Jabado
- Ester A. Serrão

Correction: Population structure and history of North Atlantic Blue whales ( Balaenoptera musculus musculus ) inferred from whole genome sequence analysis
- Sushma Jossey
- Oliver Haddrath
- Mark D. Engstrom
Genomic data identify genetic structure in Enoploctenus cyclothorax (Araneae: Ctenidae), revealing two distinct taxonomic units in the southern region of the Brazilian Atlantic Forest
- Mariana Costa Terra
- Antonio Domingos Brescovit
- Renata da Rosa

The importance of understanding clonal structure for species listing and recovery: case studies from the rare oconee bells ( Shortia brevistyla and Shortia galacifolia ; Diapensiaceae) and the federally endangered bunched arrowhead ( Sagittaria fasciculata ; Alismataceae)
- Lauren Eberth
- Ashley B. Morris

Remnant kenngoor ( Phascogale calura ) retain genetic connectivity and genetic diversity in a highly fragmented landscape
- Rhiannon S. J. de Visser
- Michelle Hall
- Renee A. Catullo

Effective population size of adult and offspring cohorts as a genetic monitoring tool in two stand-forming and wind-pollinated tree species: Fagus sylvatica L. and Picea abies (L.) Karst.
- Heike Liesebach
- Pascal Eusemann
- Barbara Fussi

Sampling through space and time: multi-year analysis reveals dynamic population genetic patterns for an amphibian metapopulation
- Chloe E. Moore
- Meryl C. Mims

Towards a genomic resolution of the Phengaris alcon species complex
- Lucas Blattner
- Goran Dušej

Genetic diversity and inbreeding in an endangered island-dwelling parrot population following repeated population bottlenecks
- Daniel Gautschi
- Robert Heinsohn
- Linda Neaves

Monitoring genome-wide diversity over contemporary time with new indicators applied to Arctic charr populations
- Sara Kurland
- Linda Laikre

Using genomic data to estimate population structure of Gopher Tortoise ( Gopherus polyphemus ) populations in Southern Alabama
- Alexander R. Krohn
- Jeffrey M. Goessling

Using recent genetic history to inform conservation options of two Lesser Caymans iguana ( Cyclura nubila caymanensis ) populations
- Thea F. Rogers
- Ewan H. Stenhouse
- Pablo Orozco-terWengel

Cross ocean-basin population genetic dynamics in a pelagic top predator of high conservation concern, the oceanic whitetip shark, Carcharhinus longimanus
- Cassandra L. Ruck
- Mahmood S. Shivji
- Andrea M. Bernard

Population genetic structure of Morelet’s and American crocodiles in Belize: hybridization, connectivity and conservation
- Clare J. Wilkie
- Marisa Tellez
- Martin J. Genner

Integrating genomics into the genetic management of the endangered mountain yellow-legged frog
- Cynthia C. Steiner
- Leah Jacobs
- Debra M. Shier

Population structure and history of North Atlantic Blue whales ( Balaenoptera musculus musculus ) inferred from whole genome sequence analysis

Population genomics of the ‘rediscovered’ threatened New Zealand storm petrel ( Fregetta maoriana ) support a single breeding colony
- Anika N. Correll Trnka
- Chris P. Gaskin
- Anna W. Santure

Clonal distribution and spatial genetic structure of the reef-building coral Galaxea fascicularis
- Yuichi Nakajima
- Patricia H. Wepfer
- Satoshi Mitarai

Conservation genomics of an endangered floodplain dragonfly, Sympetrum pedemontanum elatum (Selys), in Japan
- Wataru Higashikawa
- Mayumi Yoshimura
- Kaoru Maeto

Distinct spatial patterns of genetic structure and diversity in the butterfly Marbled White (Melanargia galathea) inhabiting fragmented grasslands
- Evelyn Terzer
- Felix Gugerli

Phylogeographic analysis points toward invasion of the Timanfaya National Park (Lanzarote; Canary Islands) by a translocated native plant ( Rumex lunaria )
- Mario A. González Carracedo
- Mariano Hernández Ferrer
- José A. Pérez Pérez

What mandrills leave behind: using fecal samples to characterize the major histocompatibility complex in a threatened primate
- Jackie Lighten
- Nicola Anthony

Contemporary reproductive patterns of Snake River Oncorhynchus nerka in Pettit Lake
- Kendra R. Eaton
- Kurt A. Tardy
- Rebecca M. Croy

Genome-wide analysis of the harbour porpoise ( Phocoena phocoena ) indicates isolation-by-distance across the North Atlantic and potential local adaptation in adjacent waters
- Marijke Autenrieth
- Katja Havenstein
- Ralph Tiedemann

Evolutionary history of the salt marsh harvest mouse mitogenome is concordant with ancient patterns of sea level rise
- Cody M. Aylward
- Laureen Barthman-Thompson
- Mark J. Statham

Changes in the spatio-temporal genetic structure of Baltic sea trout ( Salmo trutta L.) over two decades: direct and indirect effects of stocking
- Oksana Burimski
- Anti Vasemägi

Michael (Mike) William Bruford (6th June 1963–13th April 2023)
- Benoit Goossens

A comprehensive strategy for the conservation of forest tree genetic diversity: an example with the protected Pinus nigra subsp. salzmannii (Dunal) Franco in France
- C. Scotti-Saintagne
- A. de Sousa Rodrigues

Correction: Beyond Bonferroni: less conservative analyses for conservation genetics
- Shawn R. Narum
Combining genomic and field analyses to reveal migratory status in a burrowing owl population
- Lynne A. Trulio
- Debra A. Chromczak
- Kristen Ruegg

Conservation implications of diverse demographic histories: the case study of green peafowl ( Pavo muticus , Linnaeus 1766)
- Ajinkya Bharatraj Patil
- Nagarjun Vijay

Rivers have shaped the phylogeography of a narrowly distributed cycad lineage in Southwest China
- Yi-Qing Wang
- Xiu-Yan Feng

Conservation genomics of Dioon holmgrenii (Zamiaceae) reveals a history of range expansion, fragmentation, and isolation of populations
- Brian L. Dorsey
- Silva H. Salas-Morales
- Timothy J. Gregory

Comparative population genetics of habitat-forming octocorals in two marine protected areas: eco-evolutionary and management implications
- Mathilde Horaud
- Rosana Arizmendi-Meija
- Jean-Baptiste Ledoux

Revisiting conservation units for the endangered mountain yellow-legged frog species complex ( Rana muscosa , Rana sierrae ) using multiple genomic methods

Exploring genetic diversity and population structure of the Little Tern ( Sternula albifrons ) in Taiwan based on mtDNA and ddRAD sequencing data
- Mei Shuet Kong
- Chung-Hang Hung
- Wei-Jen Chen

Introgressive hybridization levels of Tilapiine species in Lake Victoria basin, Kenya inferred from microsatellite and mitochondrial DNA genotyping based on next-generation sequencing
- Gerald Kwikiriza
- Thapasya Vijayan
- Harald Meimberg

Conservation genomics of the threatened Trispot Darter ( Etheostoma trisella )
- Kayla M. Fast
- Brook L. Fluker
- Michael W. Sandel

Connectivity patterns of bottlenose dolphins ( Tursiops truncatus ) in the north-east Mediterranean: implications for local conservation
- Stefania Gaspari
- Charlotte Dooley
- Andre E. Moura

- Find a journal
- Publish with us
- Track your research
Frontiers for Young Minds
- Download PDF
What Is Genetic Diversity and Why Does it Matter?

All living things on Earth contain a unique code within them, called DNA. DNA is organised into genes, similar to the way letters are organised into words. Genes give our bodies instructions on how to function. However, the exact DNA code is different even between individuals within the same species. We call this genetic diversity. Genetic diversity causes differences in the shape of bird beaks, in the flavours of tomatoes, and even in the colour of your hair! Genetic diversity is important because it gives species a better chance of survival. However, genetic diversity can be lost when populations get smaller and isolated, which decreases a species’ ability to adapt and survive. In this article, we explore the importance of genetic diversity, discuss how it is formed and maintained in wild populations, how it is lost and why that is dangerous, and what we can do to conserve it.
Why is Everything and Everyone A Little Bit Different?
Earth contains millions of different species that all look different from one another. While some species look more similar to each other than others, like lions and tigers, they will still have differences between them. Even within each species, individuals look similar to each other but they are not identical. These differences and similarities are because of many small differences between individuals’ genes . All organisms have DNA and each individual’s DNA is organised into genes. These contain the instructions to build our bodies. This is similar to the way that letters are combined to make words that then make a story. DNA can be seen as the letters, genes the words, and their instructions are the story. Small differences in DNA might change blue eyes to green, or a butterfly’s wings from black to white, like how a word can change when you replace a letter.
The combined differences in the DNA of all individuals in a species make up the genetic diversity of that species. Genetic diversity causes individuals to have different characteristics, which we can see even in our groceries. Although all tomatoes belong to the same species, the tomatoes we eat are hugely diverse, ranging from giant beefeater tomatoes to tiny cherry tomatoes. There are also hundreds of apple varieties ( Figure 1 ), that range from red to green, tart to sweet, and some apples even have pink flesh inside! Genetic diversity is what makes these types of tomatoes and apples look so different [ 1 ]. Genetic diversity is also seen in animals. For example, dogs can be large enough to pull sledges or small enough to sit nicely on your lap. All dogs are from the same species, but they look different because of genetic diversity! Though often more difficult to see, genetic diversity is also extremely important in wild animals and plants.

- Figure 1 - An example of genetic diversity in the food we eat.
- All these apples are one species. Different alleles of the genes that control their colour cause the apples to be green, yellow, red, or almost purple. Differences in the alleles that control flavour make each type taste different.
How is Genetic Diversity Generated?
Changes to an individual’s DNA are called mutations ( Figure 2 ). Mutations can arise when mistakes are made while cells are copying DNA, like making a spelling mistake when copying a word. These mutations make up a species’ genetic diversity. Over generations, more and more mistakes are made, leading to more mutations. Most mutations are either harmful or have no impact at all, but sometimes these mutations can cause changes that are helpful for a species. The individuals that have these helpful mutations might have greater chances of survival, and have more babies as a result [ 2 ]. This is adaptation . When a mother and a father have babies, the DNA of their baby is a mix of the parents’ DNA. Babies have two copies of every gene in their DNA, one from each parent. Copies of the same gene with different mutations are called alleles . When parents make a sperm or an egg, alleles in each parent are shuffled and recombined, and only one allele of a gene ends up in each sperm or egg cell. When the reshuffled alleles from a mother and a father are combined when sperm and eggs join, new mixes of alleles are created in the babies [ 2 , 3 ]. The mixing of alleles allows for new combinations of mutations and characteristics, adding to a species’ genetic diversity ( Figure 2 ).

- Figure 2 - (A) Genetic diversity is generated when mutations create new alleles over time.
- Mixing alleles from parents creates new combinations of alleles in their babies. Organisms that can clone themselves, like bacteria, can pass alleles to each other. Each coloured dot represents a different allele. (B) Genetic diversity can be lost when habitat loss divides populations or when buildings or highways isolate populations. (C) Creating protected areas where individuals from different populations can migrate and spread their genes can help a species to maintain its genetic diversity.
Not all species need a mother and a father to make a baby. Bacteria can clone themselves ( Figure 2 ) and directly pass their alleles from a parent to its identical clone [ 3 ]. Any mistakes in the parent’s DNA will be passed on to the clone. Amazingly, bacteria can also give alleles to each other, even if they are not related! This is a unique way simple species like bacteria can increase their genetic diversity, without relying on the mixing of alleles between a mother and a father [ 4 ].
Why is Genetic Diversity Important?
When a species has a lot of differences in its DNA, we say that genetic diversity is high [ 2 ]. In species with high genetic diversity, there are lots of mutations in the DNA, which cause differences in the way individuals look as well as differences in important traits that we cannot see [ 2 ]. This is called adaptation . For example, some types of apples can grow better in hotter environments, thanks to their genes. The variety of characteristics in species with high genetic diversity means they are more likely to successfully cope with changes in their environment. A great example of this is seen in the peppered moths during the industrial revolution [ 4 ]. Natural genetic diversity in peppered moths produced different wing colours, ranging from light to dark. Before the Industrial Revolution, peppered moths with light wings were more common because they had the best camouflage on white tree trunks. The Industrial Revolution caused a lot of air pollution that started to cover tree trunks, making them black. Light-winged moths were no longer camouflaged and were easy prey for birds. But dark-winged individuals were now hidden! This meant that dark moths had an advantage and were more likely to live long enough to have babies. The babies of dark moths were also dark because of the alleles they inherited from their parents, so they were also more likely to survive. The dark moths had higher fitness and became more common as a result [ 4 ].
What Happens When Genetic Diversity is Low?
When few mutations are found in the DNA of a species, genetic diversity is said to be low [ 2 ]. Low genetic diversity means that there is a limited variety of alleles for genes within that species and so there are not many differences between individuals. This can mean that there are fewer opportunities to adapt to environmental changes. Low genetic diversity often occurs due to habitat loss. For example, when a species’ habitat is destroyed or broken up into small pieces, populations become small. Small, fragmented populations can lead to loss of genetic diversity because fewer individuals can survive in the remaining habitat so fewer individuals breed to pass on their alleles. In small populations, the choice of mates is also limited. Over time, individuals will all become related and will be forced to mate with relatives. This is inbreeding . Inbred animals often have two identical alleles for their genes because the same gene was passed on from both parents. If this allele has harmful mutations, an inbred baby can be unhealthy. This is called inbreeding depression [ 2 ].
If genetic diversity gets too low, species can go extinct and be lost forever. This is due to the combined effects of inbreeding depression and failure to adapt to change. In such cases, the introduction of new alleles can save a population. This is called genetic rescue [ 2 ]. In the 1990s conservation scientists had to use genetic rescue to save the Florida panther, which was threatened by extinction due to low genetic diversity ( Figure 3 ) [ 5 ]. Very few Florida panthers remained and their genetic diversity was extremely low. Many Florida panther babies were sick because of inbreeding depression. A closely related panther with high genetic diversity was present in Texas. Texan panthers were moved to Florida to have babies with the Florida panthers. This increased genetic diversity because of the mixing of alleles we spoke about before. Soon after the Texan panthers arrived, many healthy kittens were born [ 5 ].

- Figure 3 - (A) The Florida panther was once widespread, with high genetic diversity.
- (B) Hunting and habitat loss reduced population size and resulted in very low genetic diversity and inbreeding. (C) Eight female panthers from Texas were moved to Florida to breed with Florida panthers. (D) When the Texas and Florida panthers bred, new alleles were introduced into the population, helping the Florida panther population become bigger and healthier over time.
What’s Happening to Genetic Diversity Around the World?
We hear a lot about the loss of species in the world, but we are also seeing a loss of genetic diversity within species. The increasing number of people on Earth and our increasing use of natural resources has reduced space and resources for wild species. Over time, many wild animal and plant populations have become smaller or more isolated. Many species have also gone through local extinctions. This has led to a global loss of genetic diversity. Scientists think that the genetic diversity within species may have declined by as much as 6% globally since the Industrial Revolution [ 6 ]. This means that many species are less able to adapt when facing new challenges, like climate change, pollution, and new diseases. If too much genetic diversity is lost, more and more species could become unhealthy and in need of conservation actions similar to the Florida panther. However, there are steps we can take to conserve and restore genetic diversity across many species.
How Do We Stop Genetic Diversity Loss?
We must preserve and protect genetic diversity. This can be done through the conservation of our remaining wild populations [ 2 ]. We can use nature reserves and wildlife bridges to reconnect wild populations that have become separated by our cities and highways. We can also restore habitats, because this will allow wild populations to get bigger. Sometimes we can even remove harmful stressors and pests so that populations can naturally regrow. We can also reintroduce species that have been lost from habitats they used to live in. Taken together, these strategies can help stop genetic diversity loss. It is important to protect genetic diversity because it is the foundation for healthy species. Healthy species are necessary for human health and for the health of the whole planet!
Gene : ↑ A section of DNA that contains the instructions for a trait.
Genetic Diversity : ↑ The overall diversity in the DNA between the individuals of a species.
Mutation : ↑ A change in an organism’s DNA. This can be a change of a single letter or a much bigger change of hundreds of letters at once.
Adaptation : ↑ The process of a species changing in order to better survive in its environment.
Alleles : ↑ Different variations of a gene caused by mutations. Many species have two alleles for every gene, one copy from each parent.
Inbreeding : ↑ Breeding between closely related individuals. Inbreeding often happens when populations are small and there are few options for mating. Inbred individuals are usually less healthy.
Inbreeding Depression : ↑ Inbred individuals share ancestors and are more likely to have identical copies of genes. If these genes contain harmful mutations, they will be expressed and cause lower health of inbred individuals.
Genetic Rescue : ↑ A conservation strategy, new individuals are moved into a population to increase genetic diversity and improve population health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
[1] ↑ Meyer, R., and Purugganan, M. 2013. Evolution of crop species: genetics of domestication and diversification. Nat. Rev. Genet. 14:840–52. doi: 10.1038/nrg3605
[2] ↑ Frankham, R., Ballou, J. D., and Briscoe, D. A. 2002. Introduction to Conservation Genetics. Cambridge: Cambridge University Press. p. 617.
[3] ↑ Emamalipour M., Seidi K., Zununi V. S., Jahanban-Esfahlan A., Jaymand M., Majdi H., et al. 2020. Horizontal gene transfer: from evolutionary flexibility to disease progression. Front. Cell. Dev. Biol. 8:229. doi: 10.3389/fcell.2020.00229
[4] ↑ Cook, L. M., and Saccheri, I. J. 2013. The peppered moth and industrial melanism: evolution of a natural selection case study. Heredity 110:207–12. doi: 10.1038/hdy.2012.92
[5] ↑ Johnson, W. E., Onorato, D. P., Roelke, M. E., Land, E. D., Cunningham, M., Belden, R. C., et al. 2010. Genetic restoration of the Florida panther. Science . 329:1641–5. doi: 10.1126/science.1192891
[6] ↑ Leigh, D. M., Hendry, A. P., Vázquez-Domínguez, E., and Friesen, V. L. 2019. Estimated six per cent loss of genetic variation in wild populations since the industrial revolution. Evol. Appl. 12:1505–12. doi: 10.1111/eva.12810
Republish This Story
Genetics Studies Have a Diversity Problem That Researchers Struggle To Fix

CHARLESTON, S.C. — When he recently walked into the dental clinic at the Medical University of South Carolina donning a bright-blue pullover with “In Our DNA SC” embroidered prominently on the front, Lee Moultrie said, two Black women stopped him to ask questions.
“It’s a walking billboard,” said Moultrie, a health care advocate who serves on the community advisory board for In Our DNA SC, a study underway at the university that aims to enroll 100,000 South Carolinians — including a representative percentage of Black people — in genetics research. The goal is to better understand how genes affect health risks such as cancer and heart disease.
Moultrie, who is Black and has participated in the research project himself, used the opportunity at the dental clinic to encourage the women to sign up and contribute their DNA. He keeps brochures about the study in his car and at the barbershop he visits weekly for this reason. It’s one way he wants to help solve a problem that has plagued the field of genetics research for decades: The data is based mostly on DNA from white people.
Project leaders in Charleston told KFF Health News in 2022 that they hoped to enroll participants who reflect the demographic diversity of South Carolina, where just under 27% of residents identify as Black or African American. To date, though, they’ve failed to hit that mark. Only about 12% of the project’s participants who provided sociodemographic data identify as Black, while an additional 5% have identified as belonging to another racial minority group.
“We’d like to be a lot more diverse,” acknowledged Daniel Judge, principal investigator for the study and a cardiovascular genetics specialist at the Medical University of South Carolina.
Lack of diversity in genetics research has real health care implications. Since the completion more than 20 years ago of the Human Genome Project, which mapped most human genes for the first time, close to 90% of genomics studies have been conducted using DNA from participants of European descent, research shows . And while human beings of all races and ancestries are more than 99% genetically identical, even small differences in genes can spell big differences in health outcomes.

“Precision medicine” is a term used to describe how genetics can improve the way diseases are diagnosed and treated by considering a person’s DNA, environment, and lifestyle. But if this emerging field of health care is based on research involving mostly white people, “it could lead to mistakes, unknowingly,” said Misa Graff, an associate professor in epidemiology at the University of North Carolina and a genetics researcher.
In fact, that’s already happening. In 2016, for example, research found that some Black patients had been misdiagnosed with a potentially fatal heart condition because they’d tested positive for a genetic variant thought to be harmful. That variant is much more common among Black Americans than white Americans, the research found, and is considered likely harmless among Black people. Misclassifications can be avoided if “even modest numbers of people from diverse populations are included in sequence databases,” the authors wrote.
The genetics research project in Charleston requires participants to complete an online consent form and submit a saliva sample, either in person at a designated lab or collection event or by mail. They are not paid to participate, but they do receive a report outlining their DNA results. Those who test positive for a genetic marker linked to cancer or high cholesterol are offered a virtual appointment with a genetics counselor free of charge.
Some research projects require more time from their volunteers, which can skew the pool of participants, Graff said, because not everyone has the luxury of free time. “We need to be even more creative in how we obtain people to help contribute to studies,” she said.
Email Sign-Up
Subscribe to KFF Health News' free Morning Briefing.
Moultrie said he recently asked project leaders to reach out to African American media outlets throughout the Palmetto State to explain how the genetics research project works and to encourage Black people to participate. He also suggested that when researchers talk to Black community leaders, such as church pastors, they ought to persuade those leaders to enroll in the study instead of simply passing the message along to their congregations.
“We have new ideas. We have ways we can do this,” Moultrie said. “We’ll get there.”
Other ongoing efforts are already improving diversity in genetics research. At the National Institutes of Health, a program called “All of Us” aims to analyze the DNA of more than 1 million people across the country to build a diverse health database. So far, that program has enrolled more than 790,000 participants. Of these, more than 560,000 have provided DNA samples and about 45% identify as being part of a racial or ethnic minority group.
“Diversity is so important,” said Karriem Watson, chief engagement officer for the All of Us research program . “When you think about groups that carry the greatest burden of disease, we know that those groups are often from minoritized populations.”
Diverse participation in All of Us hasn’t come about by accident. NIH researchers strategically partnered with community health centers, faith-based groups, and Black fraternities and sororities to recruit people who have been historically underrepresented in biomedical research.
In South Carolina, for example, the NIH works with Cooperative Health, a network of federally qualified health centers near the state capital that serve many patients who are uninsured and Black, to recruit patients for All of Us. Eric Schlueter, chief medical officer of Cooperative Health, said the partnership works because their patients trust them.
“We have a strong history of being integrated into the community. Many of our employees grew up and still live in the same communities that we serve,” Schlueter said. “That is what is part of our secret sauce.”
So far, Cooperative Health has enrolled almost 3,000 people in the research program, about 70% of whom are Black.
“Our patients are just like other patients,” Schlueter said. “They want to be able to provide an opportunity for their children and their children’s children to have better health, and they realize this is an opportunity to do that.”
Theoretically, researchers at the NIH and the Medical University of South Carolina may be trying to recruit some of the same people for their separate genetics studies, although nothing would prevent a patient from participating in both efforts.

The researchers in Charleston acknowledge they still have work to do. To date, In Our DNA SC has recruited about half of the 100,000 people it hopes for, and of those, about three-quarters have submitted DNA samples.
Caitlin Allen, a program investigator and a public health researcher at the medical university, acknowledged that some of the program’s tactics haven’t succeeded in recruiting many Black participants.
For example, some patients scheduled to see providers at the Medical University of South Carolina receive an electronic message through their patient portal before an appointment, which includes information about participating in the research project. But studies show that racial and ethnic minorities are less likely to engage with their electronic health records than white patients, Allen said.
“We see low uptake” with that strategy, she said, because many of the people researchers are trying to engage likely aren’t receiving the message.
The study involves four research coordinators trained to take DNA samples, but there’s a limit to how many people they can talk to face-to-face. “We’re not necessarily able to go into every single room,” Allen said.
That said, in-person community events seem to work well for enrolling diverse participants. In March, In Our DNA SC research coordinators collected more than 30 DNA samples at a bicentennial event in Orangeburg, South Carolina, where more than 60% of residents identify as Black. Between the first and second year of the research project, Allen said, In Our DNA SC doubled the number of these community events that research coordinators attended.
“I would love to see it ramp up even more,” she said.
Related Topics
- Health Industry
- Race and Health
- Disparities
- South Carolina
Copy And Paste To Republish This Story
By Lauren Sausser April 25, 2024
CHARLESTON, S.C. — When he recently walked into the dental clinic at the Medical University of South Carolina donning a bright-blue pullover with “In Our DNA SC” embroidered prominently on the front, Lee Moultrie said, two Black women stopped him to ask questions.
“It’s a walking billboard,” said Moultrie, a health care advocate who serves on the community advisory board for In Our DNA SC, a study underway at the university that aims to enroll 100,000 South Carolinians — including a representative percentage of Black people — in genetics research. The goal is to better understand how genes affect health risks such as cancer and heart disease.
Moultrie, who is Black and has participated in the research project himself, used the opportunity at the dental clinic to encourage the women to sign up and contribute their DNA. He keeps brochures about the study in his car and at the barbershop he visits weekly for this reason. It’s one way he wants to help solve a problem that has plagued the field of genetics research for decades: The data is based mostly on DNA from white people.
Project leaders in Charleston told KFF Health News in 2022 that they hoped to enroll participants who reflect the demographic diversity of South Carolina, where just under 27% of residents identify as Black or African American. To date, though, they’ve failed to hit that mark. Only about 12% of the project’s participants who provided sociodemographic data identify as Black, while an additional 5% have identified as belonging to another racial minority group.
“We’d like to be a lot more diverse,” acknowledged Daniel Judge, principal investigator for the study and a cardiovascular genetics specialist at the Medical University of South Carolina.
“Precision medicine” is a term used to describe how genetics can improve the way diseases are diagnosed and treated by considering a person’s DNA, environment, and lifestyle. But if this emerging field of health care is based on research involving mostly white people, “it could lead to mistakes, unknowingly,” said Misa Graff, an associate professor in epidemiology at the University of North Carolina and a genetics researcher.
In fact, that’s already happening. In 2016, for example, research found that some Black patients had been misdiagnosed with a potentially fatal heart condition because they’d tested positive for a genetic variant thought to be harmful. That variant is much more common among Black Americans than white Americans, the research found, and is considered likely harmless among Black people. Misclassifications can be avoided if “even modest numbers of people from diverse populations are included in sequence databases,” the authors wrote.
Some research projects require more time from their volunteers, which can skew the pool of participants, Graff said, because not everyone has the luxury of free time. “We need to be even more creative in how we obtain people to help contribute to studies,” she said.
“We have new ideas. We have ways we can do this,” Moultrie said. “We’ll get there.”
Other ongoing efforts are already improving diversity in genetics research. At the National Institutes of Health, a program called “All of Us” aims to analyze the DNA of more than 1 million people across the country to build a diverse health database. So far, that program has enrolled more than 790,000 participants. Of these, more than 560,000 have provided DNA samples and about 45% identify as being part of a racial or ethnic minority group.
“Diversity is so important,” said Karriem Watson, chief engagement officer for the All of Us research program . “When you think about groups that carry the greatest burden of disease, we know that those groups are often from minoritized populations.”
Diverse participation in All of Us hasn’t come about by accident. NIH researchers strategically partnered with community health centers, faith-based groups, and Black fraternities and sororities to recruit people who have been historically underrepresented in biomedical research.
“We have a strong history of being integrated into the community. Many of our employees grew up and still live in the same communities that we serve,” Schlueter said. “That is what is part of our secret sauce.”
“Our patients are just like other patients,” Schlueter said. “They want to be able to provide an opportunity for their children and their children’s children to have better health, and they realize this is an opportunity to do that.”
Caitlin Allen, a program investigator and a public health researcher at the medical university, acknowledged that some of the program’s tactics haven’t succeeded in recruiting many Black participants.
“We see low uptake” with that strategy, she said, because many of the people researchers are trying to engage likely aren’t receiving the message.
The study involves four research coordinators trained to take DNA samples, but there’s a limit to how many people they can talk to face-to-face. “We’re not necessarily able to go into every single room,” Allen said.
“I would love to see it ramp up even more,” she said.
We encourage organizations to republish our content, free of charge. Here’s what we ask:
You must credit us as the original publisher, with a hyperlink to our kffhealthnews.org site. If possible, please include the original author(s) and KFF Health News” in the byline. Please preserve the hyperlinks in the story.
It’s important to note, not everything on kffhealthnews.org is available for republishing. If a story is labeled “All Rights Reserved,” we cannot grant permission to republish that item.
Have questions? Let us know at KHNHelp@kff.org
More From KFF Health News

Medical Residents Are Increasingly Avoiding States With Abortion Restrictions

KFF Health News' 'What the Health?': Newly Minted Doctors Are Avoiding Abortion Ban States

Paid Sick Leave Sticks After Many Pandemic Protections Vanish

Three People Shot at Super Bowl Parade Grapple With Bullets Left in Their Bodies
Thank you for your interest in supporting Kaiser Health News (KHN), the nation’s leading nonprofit newsroom focused on health and health policy. We distribute our journalism for free and without advertising through media partners of all sizes and in communities large and small. We appreciate all forms of engagement from our readers and listeners, and welcome your support.
KHN is an editorially independent program of KFF (Kaiser Family Foundation). You can support KHN by making a contribution to KFF, a non-profit charitable organization that is not associated with Kaiser Permanente.
Click the button below to go to KFF’s donation page which will provide more information and FAQs. Thank you!
- Search Menu
- Chemical Biology and Nucleic Acid Chemistry
- Computational Biology
- Critical Reviews and Perspectives
- Data Resources and Analyses
- Gene Regulation, Chromatin and Epigenetics
- Genome Integrity, Repair and Replication
- Methods Online
- Molecular Biology
- Nucleic Acid Enzymes
- RNA and RNA-protein complexes
- Structural Biology
- Synthetic Biology and Bioengineering
- Advance Articles
- Breakthrough Articles
- Special Collections
- Scope and Criteria for Consideration
- Author Guidelines
- Data Deposition Policy
- Database Issue Guidelines
- Web Server Issue Guidelines
- Submission Site
- About Nucleic Acids Research
- Editors & Editorial Board
- Information of Referees
- Self-Archiving Policy
- Dispatch Dates
- Advertising and Corporate Services
- Journals Career Network
- Journals on Oxford Academic
- Books on Oxford Academic
Article Contents
Introduction, materials and methods, data availability, supplementary data.
- < Previous
Multiplexed in - situ mutagenesis driven by a dCas12a-based dual-function base editor
- Article contents
- Figures & tables
- Supplementary Data
Yaokang Wu, Yang Li, Yanfeng Liu, Xiang Xiu, Jiaheng Liu, Linpei Zhang, Jianghua Li, Guocheng Du, Xueqin Lv, Jian Chen, Rodrigo Ledesma-Amaro, Long Liu, Multiplexed in - situ mutagenesis driven by a dCas12a-based dual-function base editor, Nucleic Acids Research , Volume 52, Issue 8, 8 May 2024, Pages 4739–4755, https://doi.org/10.1093/nar/gkae228
- Permissions Icon Permissions
Mutagenesis driving genetic diversity is vital for understanding and engineering biological systems. However, the lack of effective methods to generate in-situ mutagenesis in multiple genomic loci combinatorially limits the study of complex biological functions. Here, we design and construct MultiduBE, a dCas12a-based multiplexed dual-function base editor, in an all-in-one plasmid for performing combinatorial in-situ mutagenesis. Two synthetic effectors, duBE-1a and duBE-2b, are created by amalgamating the functionalities of cytosine deaminase (from hAPOBEC3A or hAID*Δ ), adenine deaminase (from TadA9), and crRNA array processing (from dCas12a). Furthermore, introducing the synthetic separator Sp4 minimizes interference in the crRNA array, thereby facilitating multiplexed in-situ mutagenesis in both Escherichia coli and Bacillus subtilis . Guided by the corresponding crRNA arrays, MultiduBE is successfully employed for cell physiology reprogramming and metabolic regulation. A novel mutation conferring streptomycin resistance has been identified in B . subtilis and incorporated into the mutant strains with multiple antibiotic resistance. Moreover, surfactin and riboflavin titers of the combinatorially mutant strains improved by 42% and 15-fold, respectively, compared with the control strains with single gene mutation. Overall, MultiduBE provides a convenient and efficient way to perform multiplexed in-situ mutagenesis.

Natural genetic diversity, even just single nucleotide variation (SNV), provides unique biological functionalities ( 1 ). Creating intentional genetic diversity via mutagenesis is a key way for better understanding and engineering biological systems, which can be followed by the identification of the mutants of interest by artificial selection or screening of the genetic variants ( 2 ). Traditional methods that combine in vitro diversification and in vivo selection can be time-consuming and tedious. To overcome these limitations, various in vivo mutagenesis systems, such as phage-assisted continuous evolution (PACE) ( 3 ), OrthoRep (powered by the orthogonal DNA polymerase) ( 4 , 5 ), and MutaT7 (driven by T7 RNA polymerase fused with a cytidine deaminase) ( 6 ), have been developed. However, these methods require the insertion of a copy of the target gene into a phage or plasmid, limiting their ability to induce mutations in the original genomic loci. As a consequence, the genetic and phenotypic diversity generated often differs from that naturally occurring ( 7 ).
Traditional chemical and physical mutagenesis methods, such as ethyl methane sulfonate (EMS) or UV mutagenesis, accelerate the microbial evolution process by inducing damage to genomic DNA. However, the mutational sites are relatively random, and their identification is limited to whole-genome sequencing. The Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) systems (e.g. Cas9 and Cas12a), which can precisely recognize the target DNA with a protospacer adjacent motif (PAM) via single guide RNA (sgRNA) or CRISPR RNA (crRNA) guidance, have revolutionized the gene editing process ( 8 ). CRISPR-derived tools, coupled with specific functional domains, enable in-situ genomic mutagenesis similar to traditional chemical and physical mutagenesis, providing this process with improved target specificity. For instance, the EvolvR system has been built to diversify targeted genomic loci by fusing a nick-translating and error-prone DNA polymerase to the nickase version of Cas9 (nCas9, D10A or H840A) ( 2 ). Furthermore, cytosine base editors (CBEs) or adenine base editors (ABEs), which produce cytosine to thymine (C > T) or adenine to guanine (A > G) conversion in the genomic target, can be created by fusing cytosine deaminases or adenine deaminases to the catalytically dead Cas9 (dCas9) or nCas9 (D10A) ( 9 ). By combining both cytosine and adenine base editing activities, dual-function BE has also been constructed based on nCas9 to generate more variation ( 10–15 ). In addition, CRISPR systems enable multiplexed activities by utilizing multiple gRNAs/crRNAs to target a range of genomic loci ( 16 , 17 ), offering great potential for mutating multiple genes in combination to reveal interdependent genetic and phenotypic variations. Cas12a (or dCas12a), known for its multiplexed advantage in producing multiple crRNAs from a CRISPR array by its own RNase activity compared with Cas9 ( 17–19 ), may be more suitable for combinatorial in - situ mutagenesis by incorporating cytosine deaminases and adenine deaminases. However, this dual-function BE derived from dCas12a has not been built yet, and the multiplexed advantage was underutilized for the dCas12a-based CBEs and ABEs ( 20–25 ).
In this work, we design and construct the dCas12a-based multiplexed dual-function BE (MultiduBE), and employ it in the multiplexed in-situ mutagenesis of various biological phenotypes, aided by specific crRNA arrays (Figure 1 ). Through composition and structure optimization, synthetic effectors duBE-1a and duBE-2b possessing combinatorial activities of cytosine deaminase, adenine deaminase, and crRNA array processing are obtained. Further modifications, including replacing the promoter of the synthetic effector and engineering the crRNA array with a synthetic separator, allow MultiduBE to achieve the synchronized multiplexed in-situ mutagenesis at five different genomic loci in both Escherichia coli and Bacillus subtilis . Using B . subtilis as the host, we study the capacity of MultiduBE to enable phenotypic diversification by selecting fluorescent proteins (sYFP2, mKate, and mTagBFP2) and cell morphology determining proteins (FtsZ and MreB) as the targets. Additionally, we generate mutants conferring resistance to various antibiotics (tetracycline, rifampicin, spectinomycin, and streptomycin), including one that has never been reported before. Moreover, we create strains with improved either surfactin or riboflavin synthesis, which demonstrate the role of MultiduBE in metabolic diversity and regulation. As a conclusion, MultiduBE can produce genetic and phenotype variations through multiplexed in-situ genome mutagenesis, showing general applicability and portability in both basic and applied research.

The workflow for the design, construction, optimization and application of the dCas12a-based multiplexed dual-function base editor (MultiduBE). MultiduBE, a versatile tool for synchronized multiplexed in - situ mutagenesis, was created by amalgamating the functionalities of cytosine deaminase, adenine deaminase, and dCas12a. Through the replacement of the synthetic effector's promoter and the engineering of the crRNA array with a synthetic separator, MultiduBE demonstrates its capability to achieve simultaneous in-situ mutagenesis at five distinct genomic loci in both Escherichia coli and Bacillus subtilis . MultiduBE was employed for performing multiplexed in-situ mutagenesis, taking generic phenotypic diversity, identification of antibiotic resistance mutation, and metabolic regulation as examples.
Chemicals and reagents
The chemicals and plasmid extraction kit were obtained from Sangon Biotech (Shanghai, China). The riboflavin, FMN, and surfactin standards were bought from Yuanye Bio-Technology (Shanghai, China). T4 DNA ligase and PrimeSTAR HS DNA polymerase were obtained from Takara Biomedical Technology (Beijing, China). Taq master mix (Dye Plus) and Phanta Max Master Mix (Dye Plus) were purchased from Vazyme Biotech (Nanjing, China). Restriction enzyme Bsa I used for Golden Gate Assembly was purchased from New England Biolabs (Beijing, China). The seamless cloning kit was obtained from Beyotime Biotechnology (Shanghai, China). The DNA gel purification kit was obtained from Thermo Scientific (Waltham, USA). Oligonucleotides and genes were synthesized from GENEWIZ (Suzhou, China). For common cell culture, Luria-Bertani (LB) medium (10 g/l tryptone, 5 g/l yeast extract, and 5 g/l NaCl) was used. The following antibiotics were used for selections in E . coli : ampicillin, 100 μg/ml; kanamycin, 50 μg/ml. The following antibiotics were used for selections in B . subtilis : kanamycin, 50 μg/ml; chloromycetin, 5 μg/ml; tetracycline, 20 μg/ml; rifampicin, 50 μg/ml; spectinomycin, 50 μg/ml; streptomycin, 200 μg/ml.
Plasmids and strains
Strains and plasmids used in this study are provided in Supplementary Note S1 and Supplementary Note S2 , respectively. The plasmids constructed in this study can be acquired from the MolecularCloud plasmid repository (MC_0101390–MC_0101405, MC_0101449–MC_0101454). The primers used for plasmids and strains construction are shown in Supplementary Note S3 . Plasmid and crRNA array sequences with the necessary annotations can be downloaded using the URL shown in Supplementary Note S2 and Supplementary Note S4 . For plasmid construction, E . coli DH5α was used as the host, and the seamless cloning kit was used according to the manufacturer's instructions.
A previously reported method was used to transform the B . subtilis strains into supercompetent cells ( 26 ). The B . subtilis strain BSZRG (G00, B. subtilis 168Δ epr:: XylR - P xylA -comKS , Δ trpC2 :: trpC0 , Δ gudB :: gudB + ) ( 27 ) was used for the verification of the base editors and the generation of antibiotic-resistant strains. Strain G600 ( B. subtilis 168Δ epr:: XylR - P xylA -comKS , Δ trpC2 :: trpC0 , Δ gudB :: gudB + , aprE 0 , nprE 0 , bpr 0 , mpr 0 , nprB 0 ) ( 27 ) was used as the start host strain for riboflavin synthesis emergence. Sur-s0 ( B. subtilis G600, sfpm0 ) was constructed aided by plasmid pWLBE-gCpf1Ng-sfpM0, and used as the start host strain for surfactin synthesis regulation. Strain G00-CmYKT ( B. subtilis G00 ybbU ::Cm R - syfp2 - mkate - mtagbfp2 ) was constructed by transforming the DNA fragment with corresponding homologous arms (S1-CmYKT) into strain G00, and used for fluorescent and morphology assay.
Base editing and sequencing analysis
Before introducing the synthetic separator, the crRNA array was constructed using the SOMACA method ( 28 ). After introducing the separator, a modified two-round PCR based method was developed and applied for the assembly of the crRNA array ( Supplementary Figure S1 ). The primers used for crRNA array construction were listed in Supplementary Note S3 . The crRNA fragment in the first position of the array was produced by 1 round PCR using primers with the Bsa I adapter (Phanta Max 10 μl, 10 μM forward primer 5 μl, 10 μM reverse primer 5 μl, 30 cycles). The crRNA fragments except for the first position were generated using two-round PCR: In round-1, a long forward primer including 27-nt DR+, N23, and 32-nt Sp4 was designed and turned into a double strand with a general reverse primer complement to Sp (Phanta Max 10 μl, 10 μM forward primer 5 μl, 10 μM reverse primer 5 μl, 15 cycles); In round-2, the general forward primer targeting DR + with a Bsa I adapter and the general reverse primer targeting Sp with a Bsa I adapter were used, with PCR product of round-1 as the template (Phanta Max 10 μl, 10 μM forward primer 5 μl, 10 μM reverse primer 5 μl, PCR product of round-1 1 μl, 30 cycles). These PCR products containing crRNA fragments were diluted 10 times and directly used for the construction of the crRNA array by Golden Gate assembly ( 18 ). Based on this method, only the long forward primer in round-1 needed to be changed if new targets will be edited.
When the crRNA array was ligated in the all-in-one BE plasmid, the generated plasmid was verified by sequencing, transformed into B . subtilis strain G00 and cultured in a LB-kanamycin agar plate at 30°C. A single colony was then picked from the plate and inoculated in a 14-ml sterile tube containing 2 ml LB-kanamycin media for pre-culture about 10 h, before being inoculated in other three parallel 14-ml sterile tubes containing 2 ml LB-kanamycin media with corresponding inducer (1 mM IPTG or 0.5 μM aTC). After 24 h induction, the targets on the genome of the 3 parallel cultures were amplified separately for analysis. For the resolution of the genome editing results, the Sanger sequencing method shown in Supplementary Figure S2A or the next-generation sequencing (NGS) method shown in Supplementary Figure S2B was employed. For Sanger sequencing, the target genes on the genome were amplified by using 2 μl culture from each of the 3 parallel as the template, and the BEAT software was employed to calculate the mutation rates ( 29 ). For high-throughput targeted amplicon sequencing, 4 fragments containing the 5 targets on aprE and nprE were amplified and equal mole mixed for sequencing, which was conducted for each of the 3 parallel cultures reads were used for each sam. In addition, potential off-target sites (POTs) were identified by the Cas-OFFinder algorithm ( 30 ) and subsequently amplified for NGS analysis. Library construction, sequencing, and data acquisition were performed on the Illumina NovaSeq platform by GENEWIZ (Suzhou, China), and the results were processed and handled using the Geneious software (> 500000 reads were used for each sample). The primers used for generating amplicons are shown in Supplementary Note S3 . After induction, the whole genome resequencing (>500 × coverage depth), RNA-seq, and miRNA sequencing of the culture were also performed and analyzed by GENEWIZ (Suzhou, China). As shown in Supplementary Table S1 , the strain G00 possesses numerous single nucleotide variations (SNVs) when compared to the reference genome of B . subtilis 168. These variations may have arisen due to long-term spontaneous mutations in the laboratory.
Assay of fluorescence intensity
The recombinant E . coli and B. subtilis strains with the fluorescence proteins were pre-cultured in LB medium for 10 h and further inoculated into 200 μl LB medium with 1% proportion at 96-well plates (corning 3603). The 96-well plates were subsequently cultured at 37°C with shaking at 750 rpm. The GFP fluorescence (excitation, 490 nm; emission, 530 nm), mCherry fluorescence (excitation, 580 nm; emission, 610 nm), and OD 600 were measured using a microplate Multi-Mode Reader (BioTek, Cytation 3) directly ( 18 ). To calculate the relative fluorescence intensities, background OD of the medium (OD bg ) and background fluorescence of the strain without fluorescent protein expression (FP bg ) was excluded, and the equation ( 1 ) was applied as follows:
Flow cytometry assay and microscopy imaging
For the verification of MultiduBE promoted fluorescent and morphology diversification, the crRNA array YKTmf5C-Sp4 was assembled into pWLT-duBE-1a and pWLT-duBE-2b and then transformed into strain G00-CmYKT. Two single colonies were picked from the two agar plates and inoculated in a 14-ml sterile tube containing 2 ml LB media with kanamycin for pre-culture about 10 h. Subsequently, the pre-culture was inoculated in a 14-ml sterile tube containing 2 ml LB-kanamycin media without aTC and a 14-ml sterile tube containing 2 ml LB-kanamycin media with 0.5 μM aTC, respectively. The two group cultures (without induction and with induction) were analyzed by flow cytometry and Laser Scanning Confocal Microscopy (LSCM).
Flow cytometry was performed on a BD FACSArica III with BD FACSDiva software (BD Biosciences). Specifically, 1 ml cultured cells were taken and washed three times with 0.012 M PBS (pH 7.4). Then, the cells resuspended to an OD 600 of 0.3 were run at a rate of 0.5 μl/s, and the events were gated based on forward and side scatters to reduce false events ( 18 )( Supplementary Figure S3 ). After gating, at least 10 000 events were collected for the analysis of each sample. All data were exported in FCS3 format and processed using FlowJo_V10 software. LSCM was carried out by a Nikon AX-SIM S microscope (Nikon, Tokyo, Japan) fitted with a 60× oil-immersion objective. The fluorescent proteins mTagBFP2, sYFP2, and mKate were excited by 405-, 488- and 561-nm laser lines of an argon-ion laser line of a He–Ne laser, respectively. Fluorescence emissions were detected with spectral detector sets BP 429–474 for mTagBFP2, BP 500–550 for sYFP2, and BP 570–616 for mKate. Image analysis was carried out on the NIS-Elements Viewer software.
Multiplexed in-situ mutagenesis and screen method
For antibiotic resistance generation, corresponding crRNAs were designed by referencing reported mutation sites in B . subtilis or aligning the genes of B . subtilis with those of other resistant mutant hosts. The MultiduBE plasmids containing crRNA arrays tetL-rpoB-Sp4, tetL-rpsE-Sp4, tetL-mtrp-Sp4, or LBEL4C-Sp4 were transformed into strain G00. The results colonies were scraped from the agar plate and inoculated in a 50-ml sterile tube containing 10 ml LB-kanamycin media for pre-culture about 10 h, before being inoculated in a 14-ml sterile tube containing 2 ml LB-kanamycin media with 0.5 μM aTC. After induction, the mutation library was washed to remove aTC, and then inoculated into a 14-ml sterile tube containing 2 ml LB-kanamycin media with corresponding antibiotics and cultured at 30°C. The grown cells were streaked on antibiotics agar plate and cultured at 30°C for single colonies isolation and screening. The MultiduBE plasmid in the screened strain was eliminated by culturing the single colony in a 14-ml sterile tube containing 2 ml LB media without kanamycin at 50°C, followed by streaking on the LB agar plate without kanamycin and culturing at 50°C ( Supplementary Figure S4A ).
For surfactin synthesis regulation, the MultiduBE plasmids pWLT-duBE-1a and pWLT-duBE-2b containing crRNA array sur5C-Sp4 were transformed into strain G600. Two single colonies were picked from the two agar plates containing the BE plasmid and inoculated in a 14-ml sterile tube containing 2 ml LB-kanamycin media for pre-culture about 10 h, before being inoculated in a 14-ml sterile tube containing 2 ml LB-kanamycin media with 0.5 μM aTC. After induction, the mutation library was washed to remove aTC, and then spread on LB-kanamycin agar plates and cultured at 30°C for single colonies isolation. The surfactin overproducing strains were screened using a chromatic visible screening method ( 31 ), in which bromothymol blue (BTB) acts as a colour indicator and cetylpyridinium chloride (CPC) acts as a mediator, in 96-well plates (corning 3596). The difference in surfactin concentrations can be exactly reflected by a regular chromatic response from faint yellow-green to dark green and bright blue ( Supplementary Figure S4B ). The MultiduBE plasmid in the 12 screened strains Sur-s1∼s12 was also eliminated by treating at 50°C.
For riboflavin synthesis strain emergence, the MultiduBE plasmids pWLT-duBE-1a and pWLT-duBE-2b containing crRNA array rib5C-Sp4 were transformed into strain G600. Two single colonies were picked from the two agar plates containing the BE plasmid and inoculated in a 14-ml sterile tube containing 2 ml LB-kanamycin media for pre-culture about 10 h, before inoculated in a 14-ml sterile tube containing 2 ml LB-kanamycin media with 0.5 μM aTC. After induction, riboflavin-producing strains in the mutation library were screened using FADS conducted in a microfluidic devices including droplet generator, droplet injector, droplet sorter, and observing chamber ( 32 ). The cells were collected by centrifugation (10 000 × g and 4°C for 2 min), washed with fresh LB-kanamycin medium, and then diluted with LB-kanamycin medium with 30 g/l glucose to an OD 600 of 0.05. In this cell density, 10% of droplets (diameter was ∼30 μm) will contain a single cell and less than 0.5% will contain more than one cell except for the remaining empty droplets, owing to the Poisson distribution of cells in the droplets. The cells were collected in a 1-ml syringe, and the oil phase consisting of HFE 7500 (3M, USA) with 2% (v/v) Pico-Surf surfactant (5% in HFE 7500, Sphere Fluidics) was also put in another 1-ml syringe. The connection between the syringe and the microfluidic device was made using polyethylene tubing with an inner diameter of 0.38 mm and an outer diameter of 1.03 mm. The flow rates of the cell solution and spacing oil were controlled by driving the syringes with two pumps (PHD2000, Harvard, USA), and water-in-oil droplets were generated due to the shearing action of the oil phase. The generated droplets were incubated at 30°C with 220 rpm for 24 h in an inverted syringe. The incubated droplets were injected into the droplet sorter, and the fluorescence of the droplet was excited by white LED light with the corresponding filters (excitation, 495 nm; emission, 525 nm) in an inverted microscope (DMi-8, Leica). The emission fluorescent light was detected by a photomultiplier tube (PMT; H10721, Hamamatsu) and analyzed by Arduino DUE. The collected droplets were put in a 1.5-ml tube containing 100 μl demulsifier 1H,1H,2H,2H-perfluoro-1-octanol (Aladdin), and 200 μl LB-kanamycin medium. The aqueous phase containing the collected cells was spread on LB-kanamycin agar plates and cultured at 30°C. The isolated single colonies were inoculated into 96-well plates (corning 3603) for fluorescence determination (excitation, 490 nm; emission, 530 nm). The MultiduBE plasmid in the 14 screened strains Rib-s1∼s14 was also eliminated by treating at 50°C ( Supplementary Figure S4C ).
Shake flasks culture and analytical methods
The medium used for shake flask culture of riboflavin or surfactin contains 80 g/l glucose, 6 g/l urea, 12 g/l yeast extract, 6 g/l tryptone, 12.5 g/l K 2 HPO 4 ·3H 2 O, 2.5 g/l KH 2 PO 4 , and 3 g/l MgSO 4 ·7H 2 O. To prepare the seed cultures, single colonies were picked into 10 ml LB medium in 250-ml shake flasks from the LB plates with the engineered B. subtilis strains. After being cultured at 37°C and 220 rpm for 10 h, the seed cultures were further inoculated into a 50 ml fermentation medium with 5% proportion at a 250-ml baffled flask and grown at 37°C and 220 rpm for 60 h ( Supplementary Figure S4D ), and three replicates were set for each strain. To measure cell densities (absorbance at 600 nm, OD 600 ), glucose, and product (riboflavin or surfactin), 1 ml cell suspension was sampled every 12 h.
Glucose in the fermentation broth was analyzed by a glucose-lactate analyzer (M100, Shenzhen Sieman Technology Co., Ltd, Shenzhen, China). For surfactin determination, 200 μl of the fermentation culture was added into 800 μl 0.1 M phosphate buffer solution (PBS, pH 8.0). After centrifugation at 10000 × g for 2 min, the supernatant was filtered through a 0.2 μm membrane. A 10 μl aliquot was injected into an HPLC system (Agilent 1260, Santa Clara, CA, USA) equipped with a variable wavelength detector (VWD, 205 nm) and a C18 column (4.6 × 250 mm; 5 μm; Waters, Ireland)( 31 ). The mobile phases were 10% water and 90% methanol containing 0.1% trifluoroacetic acid (TFA). The total flow rate of the mobile phase was kept at 1.0 ml/min and the column temperature was kept at 40°C. The total peak area of 4 surfactin isoforms was calculated for quantification of the total concentration of surfactin according to the concentration standard curve. For analyzing the expression of the srfA gene cluster, the plasmid pWLBE-gCpf1Ng-srfA-NmC was constructed to integrate mCherry into the genomic srfA gene cluster loci of specific strains. For analyzing the expression of spoIVB , spoIVB with its promoter from Sur-s0 or Sur-s1 was fused with mCherry and generated plasmid pHT-SpoIVBs0-NmC or pHT-SpoIVBs1-NmC. Similarly, remA with its promoter from Sur-s0 or Sur-s1 was fused with sfGFP and generated plasmids pHT-RemAs0-NmC and pHT-RemAs1-NmC.
For riboflavin measurement, 200 μl of the fermentation culture was added into 800 μl of 0.05 M NaOH. After centrifugation at 10 000 × g for 2 min, the supernatant was collected and diluted with 0.1 M acetate–sodium acetate buffer (pH 4.42). Subsequently, absorption at 444 nm was measured ( 33 ). The concentration of riboflavin was calculated using the following validated standard equation: Y = (OD 444 -0.0203) × DF/0.0163 ( R 2 = 0.9997; OD 444 , the absorbance value at 444 nm; Y , the concentration of riboflavin (mg/l); DF, dilution fold; OD 444 was controlled within the range of 0.1–0.8 by dilution).
The B . subtilis cells sampled at 24 h were used for the determining activity of flavokinase encoded by ribC . The cells were collected by centrifugation (10 000 × g and 4°C for 2 min), and the pellet was washed with buffer A (0.1 M PBS (pH 7.5), 1 mM dithiothreitol, and 0.1 mM EDTA) ( 34 ). The cells were resuspended in buffer A and disrupted with ultrasonic oscillation (VCX750, Sonics, CT, USA). After centrifugation at 10 000 × g and 4°C for 10 min, an aliquot of the supernatant was directly used in the flavokinase assay, and the protein concentration of the supernatant was determined by the Bradford method. Flavokinase activity was measured in a final volume of 1 ml of 0.1 M PBS (pH 7.5) containing 50 mM riboflavin, 3 mM ATP, 15 mM MgCl 2 , and 10 mM Na 2 SO 3 ( 34 ). The mixture was preincubated at 37°C for 5 min, and the reaction was started by the addition of the enzyme. The reaction was performed at 37°C for 20 min and stopped by adding 25 uL of 1.5 M trichloroacetic acid. After centrifugation at 10 000 × g for 2 min, the supernatant was filtered through a 0.2 μm membrane. A 10 μl aliquot was injected into a high-performance liquid chromatography (HPLC) system (Agilent 1260, Santa Clara, CA, USA) equipped with a fluorescence detector (FLD; excitation, 450 nm; emission, 520 nm) and a C18 column (4.6 × 250 mm; 5 μm; Waters, Ireland). The mobile phases were 35% methanol and 65% ammonium acetate (5 mM). The total flow rate of the mobile phase was kept at 1.0 ml/min and the column temperature was kept at 25°C. One unit of flavokinase activity was defined as the amount of enzyme that produced 1nmol FMN from riboflavin and ATP per min under the assay conditions. The specific activities of different strains were compared by using the flavokinase activity and protein concentration measured above. For analyzing the expression of the pur operon, the plasmid pWLBE-gCpf1Ng-pur-NmC was constructed to integrate mCherry into the genomic pur operon loci of specific strains. Similarly, the plasmid pWLBE-gCpf1Ng-rib-NmC was constructed for analyzing the expression of the rib operon of specific strains, and the plasmid pWLBE-gCpf1Ng-zwf-NmC was built to fuse mCherry to the C-terminus of ZWF on the genome of specific strains.
Statistical analysis
All experiments were independently carried out at least three times unless otherwise specified, and the results were expressed as the mean value. Two-sided t -test in Excel (Microsoft 365) was used to perform the statistical evaluation ( P -value), and P > 0.05, P < 0.05 and P < 0.01 were presented by no significance (n.s.), * and **, respectively. The graphs of results were plotted using the OriginPro software.
Coupling cytosine or adenine deaminases with dCas12a
In previous studies, Lachnospiraceae bacterium dCas12a (dLbCas12a) was commonly used for CBE or ABE construction ( 20–24 ). Here, Francisella novicida dCas12a (dFnCas12a, D917A), which possesses a lower target requirement (PAM = TTV, V = A, G, C) than that of dLbCas12a PAM = TTTV), was chosen for the building of BE. Although FnCas12a has been reported to have lower activity compared with LbCas12a and AsCas12a ( Acidaminococcus sp.) in human cells ( 35 ), it has been widely used in bacteria ( 36 ) and plants ( 37 ). To assess the compatibility between different cytosine or adenine deaminases and dFnCas12a in B. subtilis , we developed a suite of all-in-one plasmids. These plasmids were derived from a custom-made plasmid backbone pWLBE-dCas12a-N ( Supplementary Figure S5A ), in which dCas12a expression is regulated by the isopropyl β- d -1-thiogalactopyranoside (IPTG) induced promoter P grac100 , and a crRNA array implantation region is placed downstream of a constitutive promoter P veg that with a precise transcriptional start site (commonly used for B . subtilis and also exhibits activity in E . coli ) ( 27 ). The crRNA array implantation region contains a pair of head-to-head Bsa I cleavage sites flanked by two directed repeats (DRs), which allows for the rapid assembly of the required crRNA array ( 28 ) ( Supplementary Figure S5B ). The crRNA array consists of an alternant DR and a spacer, in which the DR is specifically recognized by Cas12a and the spacer (also known as guide region) serves to guide Cas12a to the complementary target DNA strand. Additionally, a temperature-sensitive replication origin pE194ts is employed to enable the elimination of the plasmid after editing the genome. Subsequently, we introduced cytosine deaminase or adenine deaminase into the plasmid backbone for genome editing validation ( Supplementary Figure S5C ). Moreover, we also co-introduced a uracil glycosylase inhibitor (UGI), which can prevent the excision of the produced uracil (U) base by the DNA repair machinery( 9 ), with cytosine deaminase to investigate whether it could enhance the efficiency of genome editing (Figure 2A ). After transforming the plasmid containing the corresponding deaminases and crRNA array into B. subtilis , IPTG was added for induction, and the editing outcomes were analyzed through Sanger sequencing and BEAT software ( 29 ) ( Supplementary Figure S2A ).

Integration of cytosine or adenine deaminases with dCas12a. ( A ) Schematic diagram for verifying compatibility between various cytosine deaminases and dCas12a. ( B ) Editing outcomes of the dCas12a-based cytosine base editors (CBEs) on target gene aprE . ( C ) A five-member crRNA array an5C target five sites on genes aprE and nprE was constructed to guide dCas12a-based cytosine base editors (CBEs) for multiplexed editing. ( D ) Editing outcomes of the dCas12a-based CBEs on target genes aprE and nprE guided by an5C. ( E ) Schematic diagram for verifying compatibility between evolved variants of adenine deaminase TadA and dCas12a. ( F ) Editing outcomes of the dCas12a-based adenine base editors (ABEs) in target genes aprE and nprE guided by an5C. Data are presented as mean values from three independent biological replicates ( n = 3).
Cytosine deaminases, including one from the family with standard editing window (rAPOBEC1) and four from the families with wider editing windows (hAPOBEC3A, PmCDA1, hAID*Δ and tCDA1EQ) ( 9 , 38 ), were codon optimized for B . subtilis and fused to the N-terminus of dCas12a, while UGI was fused to the C-terminus (Figure 2A ). In the R-loop region generated by dCas12a, the crRNA forms a double-stranded structure with the target DNA strand through complementary base pairing, while the non-targeted single strand protospacer region can undergo C > T conversion aided by the fused cytosine deaminase (Figure 2A ). To test the efficiency, the nonessential gene aprE was selected and a crRNA targeting this gene was designed. As shown in Figure 2B , we observed C > T conversion by fusing hAPOBEC3A, PmCDA1 or hAID*Δ to dCas12a, and that the incorporation of UGI enhanced editing efficiency. Among them, pWLBE-hAPOBEC3A-dCas12a-U (U means with UGI) possess the largest editing windows (at protospacer positions C8, C14, C15), and pWLBE-hAID*Δ-dCas12a-U exhibited the strongest editing efficiency (54% at position C8). To test whether dCas12a fused with cytosine deaminase and UGI retains RNase activity, enabling the maturation of the crRNA array and the recognition of multiple targets, therefore allowing multiplexed editing, 5 targets were selected on the genes aprE and nprE , and a corresponding five-member crRNA array an5C was constructed (Figure 2C and Supplementary Figure S6A ). We transformed plasmids containing various cytosine deaminases and the crRNA array an5C into B. subtilis to explore the editing outcome. Among these, pWLBE-PmCDA1-dCas12a-U was limited to inducing C > T conversion in only 1 target on the genome, whereas pWLBE-hAPOBEC3A-dCas12a-U and pWLBE-hAID*Δ-dCas12a-U can act simultaneously in 4 and 5 targets, respectively (Figure 2D ). Other six functional CDA-like (CDAL) deaminases CDAN1∼6, which have significant sequence divergence from classical AID/APOBEC family ( 39 ), were also codon optimized for B . subtilis and fused to the N-terminus of dCas12a-UGI to perform multiplexed editing ( Supplementary Figure S6B ). Among them, pWLBE-CDAN6-dCas12a-U with LjCDAL2_1 can perform C > T conversion in target 1 and target 2 simultaneously ( Supplementary Figure S6C ). To construct multiplexed ABEs, several evolved variants of adenine deaminase TadA (TadA7.10, TadA8.17, TadA8.20, TadA8e, and Tad9) ( 40 ), which can generate A > G conversion on the non-targeted single strand protospacer region ( Supplementary Figure S6D ), were fused to the N-terminus of dCas12a, respectively (Figure 2E ). Among them, pWLBE-TadA8.20-dCas12a-N (N means without UGI), pWLBE-TadA8e-dCas12a-N, and pWLBE-TadA9-dCas12a-N can achieve A > G conversion in 2, 3 and 4 targets, respectively (Figure 2F ). Furthermore, the recently reported TadA variants (T AD AC-3.1 and TadA-dual) with both adenine and cytosine deaminase activities ( 41 , 42 ) were also incorporated into the dCas12a-based BE ( Supplementary Figure S6E ). However, C > T or A > G conversion was not observed in any of the five targets ( Supplementary Figure S6F ).
Construction and optimization of the dCas12a-based MultiduBE system
To construct the dual BE system MuitiduBE, the above screened three cytosine deaminases (hAPOBEC3A, hAID*Δ, and LjCDAL2_1) and one adenine deaminase (TadA9) were incorporated into the plasmid backbone pWLBE-dCas12a-U. Guided by the five-member crRNA array an5C, the editing outcomes were analyzed. As shown in Figure 3A , pWLBE-duBE-1a and pWLBE-duBE-2b can perform dual base editing (C > T and A > G) in 4 targets simultaneously, and the cytosine deaminases in duBE-1a and duBE-2b keep their sites preferences. Although pWLBE-duBE-3b and pWLBE-duBE-3c can also edit 4 targets, only A > G conversions were found. The reachable editing targets of other synthetic effectors were less than 4. Then, we replaced the IPTG-induced promoter P grac100 in pWLBE serial plasmids with an anhydrotetracycline (aTC)-induced promoter P tet , which has low leakage and high dynamic range in both E. coli and B . subtilis ( Supplementary Figure S7 ). As shown in Figure 3B , the reachable editing targets were improved from 4 to 5 for pWLT-TadA9-dCas12a-N and pWLT-duBE-1a. Moreover, the total conversion rate for each target of pWLT-hAID*Δ-dCas12a-U and pWLT-TadA9-dCas12a-N were both over 6%. We also analyzed the expression profiles of duBE controlled by P grac100 and P tet . As shown in Supplementary Figure S8 , an increase in expression level was observed after replacing P grac100 with P tet , and both of the systems will be turned off after removing the inducer.

Construction and optimization of the dCas12a-based MultiduBE. ( A ) Composition and structure optimization of the dCas12a-based MultiduBE. ( B ) Replacing promoter of the dCas12a-based synthetic effector in pWLBE serial plasmids generating the pWLT serial plasmids. ( C ) Engineering the crRNA array for improving the editing efficiency. ( D ) Editing outcomes on target genes aprE and nprE guided by the modified five-member crRNA arrays. ( E ) A crRNA array an5cN-Sp4 containing spacers of varying lengths (14- to 26-nt) was designed and assembled into pWLT-duBE-1a and pWLT-duBE-2b from multiplexed editing. Data are presented as mean values from three independent biological replicates ( n = 3).
In natural crRNA arrays, there is a 16- to 18-nt short separator in front of the DR ( Supplementary Figure S9A ). The separator is nonessential for Cas12a targeting, and therefore we have omitted it, only including DR and spacer in the crRNA array, as it has been done in previous studies ( Supplementary Figure S9B ) ( 43 ). However, as shown in Figure 3B , the editing activities of MultiduBE on target 5 were very low (2% for pWLT-duBE-1a and 0% for pWLT-duBE-2b). This may result from interference between adjacent crRNAs after removing the separator, as suggested in another study on CRISPR activation (CRISPRa) ( 44 ). The addition of a very short 4-nt synthetic separator to isolate interference in the crRNA array has been shown to enhance the multiplex activity of dCas12a-based CRISPRa in human cells ( Supplementary Figure S9C ) ( 44 ). Therefore, we hypothesized that introducing a synthetic separator to reduce interference between adjacent crRNAs could increase the multiplex mutation rate mediated by the dCas12a-based duBE in bacterial cells ( Supplementary Figure S9D ). For matching the two-round PCR based crRNA array construction method ( Supplementary Figure S1 ), five synthetic separators (Sp1∼Sp5), with Tm values ranging from 55 to 60°C, were generated by modifying the natural FnCas12a separator (FnSp) or assembling separators from different hosts (Figure 3C and Supplementary Figure S10A ). In addition, the DR region in the array has also been appropriately extended from 19-nt to 27-nt at the 5′-terminus (Figure 3C and Supplementary Figure S10B ) to improve the Tm value to 55°C, which does not affect the formation of the crRNA stem-loop.
For pWLT-duBE-1a, adding Sp1, Sp2, Sp3 and Sp4 into the array improved the conversion rates at position C8 in target 5 from 2% to 43, 24, 38 and 43%, respectively (Figure 3D ). As to pWLT-duBE-2b, adding Sp1, Sp3, Sp4 and Sp5 into the array improved the conversion rates at position C11 in target 5 from 0% to 9, 5, 7 and 6%, respectively (Figure 3D ). After comprehensively comparing the improvement of editing efficiencies in other targets, Sp4 was chosen as the insulator for the crRNA array of MultiduBE. Subsequently, a crRNA array an5cN-Sp4 containing spacers of varying lengths (14- to 26-nt) was also constructed (Figure 3E ). We found that a crRNA larger than 17-nt was enough to guide the MultiduBE, although the editing efficiency guided by an5cN-Sp4 was lower than that guided by an5c-Sp4 (Figure 3D ). As shown in Supplementary Figure S10C , we also built crRNA arrays an5C-Sp4t (containing Sp4 and the original 19-nt DR), an5C-SpN (containing natural FnSp and the original 19-nt DR), and an5C-SpQ (containing the short 4-nt Sp and the original 19-nt DR). Both duBE-1a and duBE-2b guided by these arrays could perform editing in the five targets, but the editing efficiencies are all lower than those guided by an5C-Sp4 ( Supplementary Figure S10D ).
We then conducted next generation sequencing (NGS) analysis of the mature RNA generated by an5C and an5C-Sp4, and found that the introduction of Sp4 indeed promoted the processing of the crRNA array, resulting in the mature crRNAs containing only DR and spacer ( Supplementary Figure S10E ). This effect was particularly noticeable for the last crRNA, which exhibited poor maturation in cn5C. As shown in Supplementary Figure S11A , by changing the order of crRNAs in an5C and an5C-Sp4, crRNA arrays an5C-R1, an5C-R2, an5C-R3, an5C-R1-Sp4, an5C-R2-Sp4, an5C-R3-Sp4 have been constructed. In addition, arrays with individual crRNA were also built. As expected, the highest base conversion rates were observed in all the targets guided by the individual crRNA. In each of the crRNA arrays, placing the crRNA in the first position could maintain its maximal activity even without Sp4, while placing it in a relatively later position required the assistance of Sp4 to maintain higher editing efficiency ( Supplementary Figure S11B ).
Whole genome resequencing was also performed after treating BSZRG (G00, B. subtilis 168Δ epr:: XylR - P xylA -comKS , Δ trpC2 :: trpC0 , Δ gudB :: gudB + ) strain ( 27 ) with MultiduBE. As shown in Supplementary Table S1 , only one low-frequency C > T or A > G conversion was found out of the target genes for pWLT-duBE-1a-an5C, pWLT-duBE-1a-an5C-Sp4, and pWLT-duBE-2b-an5C-Sp4, and the mutation found in variational genomic position indicates that the crRNA-independent off-target occurred with the overexpression of cytosine and adenine deaminases. Moreover, the higher mutation frequencies in aprE and nprE still indicate that MultiduBE has a high specific targeting activity. Editing in potential off-target sites (POTs) predicted by the Cas-OFFinder algorithm ( 30 ) was also analyzed by tNGS (>500 000 reads were used), and no base conversion was observed ( Supplementary Table S2 ). As shown in Supplementary Figure S12 , transcriptome-wide RNA-seq was also employed, and off-target RNA editing, which was reported in human cells before with the nCas9-based BEs ( 45 ), was not observed here.
Multiplexed in-situ mutagenesis mediated by MultiduBE
To evaluate the multiplexed in - situ mutagenesis ability of MultiduBE, we further analyzed the detailed editing outcomes of pWLT-duBE-1a and pWLT-duBE-2b using high-throughput targeted amplicon sequencing ( Supplementary Figure S2B ). More than 500000 reads were used for the analysis of mutagenesis in each target, and the top 10 alleles guided by an5C or an5C-Sp4 were compared ( Supplementary Table S3 ). As shown in Figure 4 , the composition of mutations produced by pWLT-duBE-1a and pWLT-duBE-2b were different. The introduction of Sp4 slightly decreased the editing efficiency in target 1 for both duBE-1a and duBE-2b, which may be a result of the increased competition with other mature crRNAs that reduced the relative proportion of dCas12a bound to the first crRNA. For target 5, adding Sp4 could significantly improve the editing efficiencies for both MultiduBEs, which was consistent with the previous results analyzed by Sanger sequencing (Figure 3D ). For example, the C8T (C > T conversion at position 8 of the protospacer) conversion in target 5 produced by pWLBE-duBE-1a was improved from 11.05 to 40.41%, and the C8T + C11T conversion in target 5 produced by pWLBE-duBE-2b was improved from 1.50 to 5.28%.

Detailed editing outcomes of the MultiduBE-promoted multiplexed in-situ mutagenesis. ( A ) High-throughput targeted amplicon sequencing (tNGS) analysis for pWLT-duBE-1a guided by an5C or an5C-Sp4. ( B ) The tNGS analysis for pWLT-duBE-2b guided by an5C or an5C-Sp4. The top 10 alleles produced from an5C and an5C-Sp4 in each target were selected and compared. The asterisk means C > T or A > G conversion occurred in the complementary DNA strand of protospacer. The alleles marked in blue mean the bystander editing out-of-protospacer (BEOP), and those with red underline mean combinatorial C > T and A > G mutants. The short lines represent mean values from three independent biological replicates ( n = 3), and the circles represent individual data points. More than 500 000 reads were used for each sample. See Supplementary Table S3 for details including the reads of NGS.
As shown in Figure 4A , when pWLT-duBE-1a was used for multiplexed editing, the conversion rate of A > G in target 1 was very low (0.87% for an5C and 0.86% for an5C-Sp4). In target 1, 8 out of the 9 mutation alleles only possess C > T conversion, and no combinatorial C > T and A > G mutation was found. In target 2 to target 5, the combinatorial C > T and A > G mutations were observed. In addition, the bystander editing out-of-protospacer (BEOP) was also found in targets 1, 3 and 5 with a lower frequency (less than 2%). For example, a C > T conversion occurred in the complementary DNA strand at position 40 of the protospacer (C40T*) in target 1 (0.31% for an5C and 0.32% an5C-Sp4). As shown in Figure 4B , when pWLT-duBE-2b was used for multiplexed editing, both the conversion rate and amount of A > G in target 1 were higher than that of pWLT-duBE-1a, but the combinatorial C > T and A > G mutation was also not found in this target. Furthermore, the combinatorial C > T and A > G mutations were only found in target 2 and target 3, and a low frequency (<0.5%) BEOP was found in target 1, target 4, and target 5.
In addition to the population-level analysis described above, mutations in the targeting genes aprE and nprE treated by two MultiduBEs (duBE-1a and duBE-2b) with an5C-Sp4 were also analyzed at the single-colony level ( Supplementary Figure S13A ). Among the 8 randomly chosen single colonies, 7 different mutant strains and 1 wild type strain were found both for pWLT-duBE-1a-an5C-Sp4 and pWLT-duBE-2b-an5C-Sp4 ( Supplementary Figure S13B, C ). In addition, duBE-1a generated a strain with combinatorial mutations in all 5 targets ( Supplementary Figure S13B ). For duBE-2b, although mutations at each target were detected, in the same colony, mutations occurred at most in only 3 targets. This aligns with the theory that the probability of combinatorial mutations occurring in the same cell becomes exceedingly low after the superposition of probabilities. These results demonstrate the efficiency and diversity of MultiduBE for multiplexed in-situ mutagenesis. To prove the universality of MultiduBEs we also tested it in E. coli , and found that pWLT-duBE-1a enables multi-target dual-function editing (C > T and A > G), and the simultaneous mutagenesis in 5 targets can be achieved after introducing Sp4 into the crRNA array xy5C ( Supplementary Figure S14 ).
Multiplexed in-situ mutagenesis for cell physiology reprogramming
As demonstrated in Figure 5A , the generic application of the MultiduBE in phenotypic diversification was verified by constructing a five-member crRNA array YKTmf5C-Sp4 targeting three fluorescent proteins (sYFP2, mKate and mTagBFP2) and two cell morphology determining proteins (FtsZ and MreB) ( 46 ). The crRNAs were designed to target the 5′-untranslated region (UTR) or N-terminal coding sequence (NCS), resulting in an expression change for these genes (Figure 5B ). As depicted in Supplementary Figure S15A , two-dimensional representations of flow cytometry revealed a more heterogeneous population, particularly in the expression of mKate and mTagBFP2, after treatment with MultiduBE. Additionally, confocal images were acquired to visualize the phenotypic changes in the cells ( Supplementary Figure S15B ). As anticipated, MultiduBE treatment produced various combinations of colors and shapes without selective pressure (Figure 5C ).

Multiplexed in-situ mutagenesis for cell physiology reprogramming. ( A ) To validate MultiduBE in generic phenotypic diversification, strain G00-CmYKT was constructed by integrating three fluorescent proteins (sYFP2, mKate, and mtagBFP2) into the genome of strain G00. For cellular morphology reprogramming, FtsZ (tubulin to form Z-ring at the midcell) and MreB (cytoskeleton protein) were selected as the targets for MultiduBE. ( B ) The target table containing designed crRNAs for verifying MultiduBE in generic phenotypic diversification. ( C ) The five-member crRNA array YKTmc5C-Sp4 guided MultiduBE in generic phenotypic diversification. After treatment by MultiduBE, confocal images of strain G00-CmYKT were obtained by merging the brightfield, green (500–550 nm), red (570–616 nm) and blue (429–474 nm) channels. ( D ) The target table containing designed crRNAs for antibiotic (tetracycline, rifampicin, spectinomycin, or streptomycin) resistant mutant generation. Nucleotides marked in red indicate potential mutation sites based on previous studies. ( E ) Sequencing analysis of the crRNA arrays and the genomic targets. ( F ) Based on the crRNA arrays enriched during the screening process, a four-member crRNA array LBEL4C-Sp4 was designed to confer resistance to the four types of antibiotics used. M1–M3 represent the corresponding mutations for each gene, and details can be found in Supplementary Figure S16C . ( G ) Approximately 10 8 cells were diluted with distilled water, and 5 μl aliquots of the cell suspension were spotted onto the agar plate with specific antibiotics. ( H ) Growth curves of the wild and mutated strains. Data are presented as mean values ± SD from three independent biological replicates (n = 3). Lines indicate the mean, and shaded areas represent the SD.
Alterations in a single nucleotide of the bacteria genome can have an extensive impact on cell phenotype ( 1 ). For instance, point mutations in specific genes are known to be linked to antibiotic resistance ( 47 ). Hence, we opted to validate the multiplexed in-situ mutagenesis activity of MultiduBE by targeting endogenous genes related to antibiotic resistance. Previous studies in B . subtilis have demonstrated that rifampicin, spectinomycin, streptomycin, and tetracycline resistance can be produced through mutations of the rpoB gene (encoding RNA polymerase beta subunit, H482R/Y) ( 48 ), rpsE gene (encoding ribosomal protein S5, K23E) ( 1 ), mthA gene (encoding methylthioadenine/S-adenosylhomocysteine nucleosidase, nonsense mutation) ( 49 ), and the promoter region of tetL gene (encoding tetracycline efflux protein leader peptide) ( 50 ), respectively. In addition, investigations in other hosts also indicated potential mutations for conferring spectinomycin and streptomycin resistance in B . subtilis ( Supplementary Figure S16A ). The ribosomal protein S5 (encoded by rpsE ) from Neisseria gonorrhoeae ( 51 , 52 ) and Streptomyces filamentosus ( 53 ) were aligned with RpsE of B . subtilis to assign potential targets for conferring spectinomycin resistance (V21, A22, K26, and R30), and ribosomal protein S12 (encoded by rpsL ) from Borrelia burgdorferi ( 54 ), Mycobacterium tuberculosis ( 55 ), and Streptomyces coelicolor ( 56 ) were aligned with RpsL of B . subtilis to assign potential targets for conferring streptomycin resistance (K55 and K101). We then designed the corresponding crRNAs (Figure 5D ).
We first constructed a two-member crRNA array targeting P tetL and rpoB to investigate whether MultiduBE could generate resistances to tetracycline and rifampicin. Furthermore, we selected 4 different crRNAs targeting the potential mutation sites in rpsE and combined them with the crRNA targeting P tetL to form a minipool library containing 4 two-member crRNA arrays, which was used to generate resistances to tetracycline and spectinomycin. We also designed 2 crRNAs in rpsL and 1 crRNA in mthA and combined them with the crRNA targeting P tetL to form a minipool library including 3 two-member crRNA arrays, which was used to generate resistances to tetracycline and streptomycin. As the mutagenesis spectrums of pWLT-duBE-1a and pWLT-duBE-2b were different, we simultaneously ligated the above two-member crRNA arrays into a mixture of pWLT-duBE-1a and pWLT-duBE-2b. The generated minipool libraries were transformed into the B . subtilis strain G00 for performing multiplexed in-situ mutagenesis, and overlapping sequencing peaks were found for the crRNA array in strains with the minipool library before selection by corresponding antibiotics (Figure 5E ). After the antibiotic screening, the crRNA arrays that confer the appropriate resistance were enriched, as indicated by the pure sequencing peaks. Accordingly, the mutations occurred in the genome were consistent with those enriched crRNA arrays (Figure 5E ). Subsequently, sequencing analysis was performed by selecting three single colonies in the three corresponding two-resistance agar plates. The three colonies with tetracycline-rifampicin resistance or tetracycline-spectinomycin resistance had the same mutations, while the three colonies with tetracycline-streptomycin resistance showed different mutations (Figure 5E and Supplementary Figure S16B ). Moreover, we have identified a novel RpsL mutation, R99C, conferring streptomycin resistance in B . subtilis . This mutation is distinct from the naturally occurring K88R/E/Q/T mutations found in B . burgdorferi , M . tuberculosis , and S . coelicolor (corresponding to K101 in B . subtilis ) or the K43R mutation in M . tuberculosis (corresponding to K55 in B . subtilis ).
Based on the crRNA arrays enriched during the screening process, we designed a four-member crRNA array LBEL4C-Sp4 to confer resistance to the four types of antibiotics used above (Figure 5F ). After conducting multiplexed in-situ mutagenesis with the guidance of LBEL4C-Sp4, one tetracycline-rifampicin-spectinomycin-streptomycin resistant strain, two tetracycline-rifampicin-streptomycin resistant strains, and three tetracycline-spectinomycin-streptomycin resistant strains were obtained (Figure 5G and Supplementary Figure S16C ). These antibiotic-resistance mutants can be used for the development and research of new antibiotics. Moreover, the fitness cost of resistance, which would allow susceptible bacteria to outcompete resistant bacteria if the selective pressure from antibiotics is reduced ( 57 ), was determined from the reduced bacterial growth rate (Figure 5H ).
Multiplexed in-situ mutagenesis for metabolic regulation
Surfactin is a lipopeptide biosurfactant synthesized by a non-ribosomal peptide synthetase (NRPS) in Bacillus species, and it is widely used in the fields of biopharmaceuticals, environmental remediation, oilfield exploitation, cosmetics, and daily necessities ( 58 ). As shown in Figure 6A , we selected five genes as the targets for surfactin synthesis overproduction based on previous studies ( 58–60 ), including srfAA (encoding surfactin synthetase, a critical speed limiting step), remA (encoding the regulatory protein of biofilm formation-related genes, associated with cell fitness), spoIVB (encoding serine protease, related to spore formation), lcfB (encoding long-chain fatty-acid-CoA ligase, providing precursor fatty acyl-CoA), and ilvB (encoding acetolactate synthase large subunit, providing precursor branched-chain amino acids). The targets for crRNAs include the negative regulation sites in the promoters of srfAA , lcfB , and ilvB ; the 5′- UTR and NCS of remA ; as well as the core region between -10 and -35 in the promoter of spoIVB (Figure 6A ).

Multiplexed in-situ mutagenesis for metabolic regulation. ( A ) The target table containing designed crRNAs for surfactin synthesis regulation. ( B ) A five-member crRNA array sur5C-Sp4 was constructed and utilized for multiplexed in-situ mutagenesis on genes related to surfactin synthesis. Using a colorimetric method, 12 mutated strains (Sur-s1∼s12) were screened. ( C ) Strains G600, Sur-s0, Sur-s1, Sur-s11 and Sur-s12 were selected for shake-flask production of surfactin. M1 represents the corresponding mutation for each gene, and details can be found in Supplementary Table S4 . Data are presented as mean values ± SD from three independent biological replicates ( n = 3). The circles represent individual data points of surfactin titer, and the squares represent individual data points of OD 600 . The chromatogram of the surfactin standard (including four surfactin isoforms) is shown in yellow with a light shadow, while the surfactin chromatograms of selected strains are shown in the colors corresponding to the circles. ( D ) The synthetic pathway and regulation manner of riboflavin in B . subtilis . ( E ) The target table containing designed crRNAs for the emergence of the riboflavin overproducer. Nucleotides marked in red indicate potential mutation sites based on previous studies. ( F ) A five-member crRNA array rib5C-Sp4 was constructed to facilitate multiplexed in-situ mutagenesis for the emergence of riboflavin overproducer from strain G600. Fluorescence-activated droplet sorting (FADS) was conducted to screen the mutations overproducing riboflavin. ( G ) The isolated single colonies were picked and screened by fluorescence measurement in 96-well plates. ( H ) Five mutated strains (Rib-s4, Rib-s8, Rib-s11, Rib-s12 and Rib-s14) were selected for shake-flask culture. The strain Rib-s0, constructed by replacing P ribD and FMN riboswitch with a strong constitutive promoter P veg , was used as a control. M1–M5 represent different mutations for each gene, and details can be found in Supplementary Table S5 . Data are presented as mean values ± SD from three independent biological replicates ( n = 3). Lines indicate the mean and shaded areas represent the SD.
The 4′-phosphopantetheinyl transferase for activating surfactin synthetase encoded by sfp is inactive in B . subtilis 168 ( 59 ), so we first performed a reverse mutation on the sfp gene in strain G600 ( B. subtilis 168Δ epr:: XylR - P xylA -comKS , Δ trpC2 :: trpC0 , Δ gudB :: gudB + , aprE 0 , nprE 0 , bpr 0 , mpr 0 , nprB 0 ) ( 27 ), generating the strain Sur-s0. Then, a five-member crRNA array sur5C-Sp4 was constructed and used for the multiplexed in-situ mutagenesis in the genes related to surfactin synthesis in the strain Sur-s0 (Figure 6B ). Based on a colorimetric method ( 31 ), 12 mutant strains (Sur-s1∼s12) were screened (Figure 6B ), and the mutations in the target genes were analyzed after eliminating the MultiduBE plasmid ( Supplementary Table S4 ). Although editing occurred at all five targets guided by sur5C-Sp4 ( Supplementary Figure S17A ), the maximum number of mutations obtained involved only three genes. Finally, strains G600, Sur-s0, Sur-s1, Sur-s11, and Sur-s12 were selected for shake-flask production of surfactin. As shown in Figure 6C , in comparison to the starting strain Sur-s0 that could synthesize 702.7 mg/l surfactin, the surfactin titer of the strain Sur-s1 (with mutations in three other genes srfAA , remA and spoIVB ) improved by 42.0% to 997.5 mg/l. Furthermore, the changes in the expression of these three mutated genes in Sur-s1 were also consistent with the improvement of surfactin production ( Supplementary Figure S17B ). The growths of these five strains were similar except for Sur-s11, which showed impaired growth and reduced surfactin titer (Figure 6C ). Given that the probability of occurrence decreases with the combination of mutations, more efficient screening methods are required for combinatorial mutations involving a greater number of genes in this situation.
Riboflavin (also known as vitamin B2) is a heat-resistant, water-soluble vitamin that is essential for humans as it is a direct precursor for the coenzymes flavin adenine (FAD) and flavin mononucleotide (FMN), which are crucial for cellular processes ( 61 ). As shown in Figure 6D , the synthesis of riboflavin in B . subtilis is subjected negative feedback regulation with GTP and FMN by the guanine and FMN riboswitches, respectively ( 61 ). In addition, riboflavin kinase, encoded by the essential gene ribC , can convert riboflavin to FMN, which not only consumes riboflavin but also further strengthens the feedback inhibition of the FMN riboswitch ( 61 ). Moreover, glucose-6-phosphate dehydrogenase, encoded by zwf , is a key step in the pentose phosphate pathway, which is also known to affect riboflavin synthesis ( 61 ). Therefore, we selected the guanine riboswitch in front of purE , the FMN riboswitch in front of ribD, ribC , and zwf as mutagenic targets for enhancing riboflavin synthesis in the wild strain G600. As shown in Figure 6E , crRNAs were designed to target the key points of the two riboswitches and RibC (marked in red), as well as the 5′-UTR and NCS of Zwf. Moreover, the crRNA targeting P tet for tetracycline resistance described earlier was set to observe the induced base editing without corresponding antibiotic selection pressure.
As shown in Figure 6F , a five-member crRNA array rib5C-Sp4 was constructed in the MultiduBE plasmids pWLT-duBE-1a and pWLT-duBE-2b to promote the multiplexed in-situ mutagenesis for the emergence of riboflavin overproducer from strain G600 that do not overproduce riboflavin. After being rewashed with medium without aTC, fluorescence-activated droplet sorting (FADS) was conducted to screen and select the mutants with high fluorescence, which correlates with a high extracellular riboflavin titer in the droplets (Figure 6F and Supplementary Figure S17C ). The cells in the collected droplets were spread on agar plates containing kanamycin, and single colonies were picked and screened by fluorescence measurement in 96-well plates (Figure 6G ). A total of 14 mutated strains (Rib-s1∼s14) with high fluorescence were obtained, and the mutations in the target genes were analyzed after eliminating the MultiduBE plasmid ( Supplementary Table S5 ). Although tetracycline was not used for selection, all 14 strains had mutations in the P tet site. The mutations observed in the selected strains were more diverse than those in the surfactin synthesis regulation, with all five targets in strain Rib-S12 undergoing mutations. This highlights the importance of an efficient screening strategy for obtaining multi-site combinatorial mutants with low probabilities. Subsequently, five mutant strains (Rib-s4, Rib-s8, Rib-s11, Rib-s12 and Rib-s14) were selected for shake-flask culture, and a control strain Rib-s0 was constructed by replacing P ribD and the FMN riboswitch with a strong constitutive promoter P veg (Figure 6H ). All five mutated strains could produce more riboflavin than that of the control strain Rib-s0 (21.5 mg/l), and the riboflavin titer of Rib-s12 (344.8 mg/l) was 16-fold that of the control (Figure 6H ). Moreover, the increased expression of the synthetic pathways (encoded by the pur operon, the rib operon, and zwf ) and the reduced enzyme activity of the riboflavin consumption pathway (encoded by ribC ) in Rib-s12 correspond with the riboflavin overproduction phenotype ( Supplementary Figure S17D ).
The ability to generate in-situ mutagenesis in multiple genomic loci combinatorially is a powerful way for the study of complex biological functions. In this study, we combined the multiplexed capabilities of dCas12a with the activities of two deaminases to create MultiduBE, which can simultaneously mutate C and A (or G and T in the opposite strand) bases at multiple targets (Figure 1 ). MultiduBE, encoded on a single plasmid, allows for multiplexed in - situ mutagenesis, relying only on trans-action guided by specific crRNA arrays and therefore it does not require further genomic manipulation. Despite the possibility of off-targeting or unexpected mutations ( Supplementary Table S1 ), the successful generation of mutant strains exhibiting desired properties (Figures 5 , 6 ) confirms the efficacy of MultiduBE in directed mutagenesis. In comparison to other transcriptional regulatory processes, such as CRISPR interference and activation (CRISPRi/a) ( 8 , 19 ), MultiduBE stands out by inducing mutations at the genome level, providing greater stability and robustness to the resulting modifications. This genome-level modification can also serve as the foundation for subsequent dynamic regulation systems based on CRISPRi/a. In contrast to the previously used nCas9-based CBE or dual-function BE integrated into the genome ( 62 , 63 ), our method employs an all-in-one plasmid with general applicability in both E . coli and B . subtilis (Figure 3 and Supplementary Figure S14 ), which ensures better genetic stability by eliminating the MultiduBE plasmid after mutagenesis. For the initial construction and optimization of MultiduBE, we employed a rapid and cost-effective analysis process using Sanger sequencing. However, Sanger sequencing cannot detect low-frequency mutations and reveal detailed components of the mutations. Therefore, we also used NGS for a more detailed mutation analysis, which further revealed the diversity of in-situ mutations generated by MultiduBE (Figure 4 ). Although we observed low-frequency BEOP, these mutations were still beneficial for diversity, as we observed in ribC for the overproduction of riboflavin ( Supplementary Table S5 ).
In previous BE works based on Cas9, the nickase nCas9 (D10A) is often employed as it improves editing efficiency by an average of threefold ( 64 ). Owning to the nick produced in the non-edited target DNA strand by nCas9, DNA replication and repair will primarily use the edited DNA strand without nick as the template. As illustrated in Supplementary Figure S18A , we investigated multiplex editing performance of the nCas9-based duBE by modifying a previously reported system ( 63 ) and compared it with the dCas12a-based duBE. The sgRNA array constructed with multiple promoters showed high instability, with the majority experiencing recombination and fragment deletion after transformation into B . subtilis ( Supplementary Figure S18B,C ). Conversely, the tRNA-based sgRNA array demonstrated relatively better stability, and fragment deletion was only observed in some of the single colonies after transformation into B . subtilis but not in cultures after induction ( Supplementary Figure S18C,D ). Although the nCas9-based duBE possesses a higher activity than that based on dCas12a, the editing efficiency was significantly reduced when incorporating these sgRNAs into the five-member tRNA-sgRNA array, especially losing editing in target 3 ( Supplementary Figure S18D ). In comparison, the stability of crRNA arrays proved significantly superior to sgRNA arrays, with no instances of fragment deletion observed in both single colonies after transformation into B . subtilis and cultures after induction ( Supplementary Figure S18C,E ). Moreover, the property of nCas9 also creates diversity base conversions except for C > T and A > G ( 65–67 ). For instance, uracil-DNA glycosylase (UNG), which will remove the intermedium U base produced by nCas9 and cytosine deaminase, can initiate base-excision repair (BER) and allows the construction of BE enabled C > A and C > G conversions ( 66 , 67 ). After replacing nCas9 with dCas9, only the C > T conversion can be found, approving the specific DNA damage caused by nCas9 is necessary for UNG-initiated C > A and C > G conversions. Similarly, the BE enabled A > C and A > T editing was also built by coupling nCas9 and adenine deaminase with an N-methylpurine DNA glycosylase (MPG), which can excise the intermedium hypoxanthine (I) base and initiates BER ( 65 ). Cas12a contrary to Cas9, possesses an RNase activity, which is beneficial for the multi-targeting of MultiduBE (Figure 1 ). However, the nickase version of Cas12a that solely cleaves the target DNA strand does not exist (the cleavage of the target strand by the Nuc domain relies on the non-target strand cleavage by the RuvC domain) ( 68 ), thereby limiting both the editing efficiency (Figure 3 and Figure 4 ) and diversity of the Cas12a-based BE. This could explain why certain TadA variants, such as TadA7.10, TadA8.17, TADAC-3.1, and TadA-dual, commonly used with nCas9, showed no activity when fused with dCas12a (Figure 2F and Supplementary Figure S6E ). In the future, engineered nickase version of Cas12a that could only cleave the target DNA strand may be built by coupling and modifying the endonuclease cleavage domain ( 69 ), which will further improve the editing efficiency of BE based on dCas12a.
Previous studies on multiplexed cytosine or adenine base editing using Cas12a focused predominantly on the synthetic BE protein ( 20–24 ), paying less attention to the impact of the crRNA array. Here, we designed relatively long separators and extended the DR as needed to match the crRNA array assembly process ( Supplementary Figure S1 ). These modifications of the crRNA array significantly increased the multi-target editing efficiency in both E. coli and B . subtilis (Figure 4 and Supplementary Figure S16 ). We also developed a two-round PCR based crRNA array assembly strategy, which requires only the synthesis of an 82-nt forward primer containing DR+, N23, and 32-nt Sp4 for editing new targets ( Supplementary Figure S1 ). This novel strategy enables the production of large-scale crRNA arrays, where an oligo pool is synthesized on a DNA chip, leading to random multi-target genome-wide mutagenesis. The mthA mutation in B . subtilis can confer low-level resistance to streptomycin ( 49 ), so we mixed the crRNA targeting mthA with crRNAs targeting rpsL , a previously validated target in other hosts ( 56 ), into a crRNA minipool library for streptomycin-resistant mutant generation (Figure 5D and Supplementary Figure S16A ). With the selective pressure, novel mutations of rpsL conferring streptomycin resistance were found for B . subtilis (Figure 5E ). The robust correlation between genomic mutations and the crRNAs implies that the crRNA array could serve as trackers for mutation targets in the genome. Additionally, for the multiplexed in-situ mutagenesis guided by the crRNA array rib5C-Sp4, the mutation in P tetL was found in all the selected riboflavin overproducing strains (Figure 6H and Supplementary Table S5 ), even when the corresponding selective pressure tetracycline was not exerted. This result suggests that tetracycline resistance can be employed to preclude unmutated strains. However, as demonstrated in metabolic regulation (Figure 6 ), an efficient screening method is also essential for acquiring specific mutations from a vast combinatorial mutant library. Recent researches in genetically encoded biosensors have provided excellent opportunities in this regard ( 70 ).
Some limitations should be considered in the context of multiplexed mutagenesis using MultiduBE. For example, the challenge in constructing crRNA arrays is increased when a large number of genes are selected as the targets simultaneously. Additionally, while MultiduBE showed no substrate motif preference, its high activity was confined to a specific editing window (protospacer positions 7–15) ( Supplementary Figure S19 ). To potentially expand the editing window, liquid-liquid phase separation (LLPS) could be explored, as it has been shown to improve the efficiency of CRISPRa in mammalian cells and mice ( 71 ). As shown in Supplementary Figure S20 , we also try to broaden the targeting scope to the non-canonical PAM (ncPAM ≠ TTV) by constructing the broad-spectrum duBE (duBEbs), incorporating a broad-spectrum variant of dCas12a ( 72 ). However, duBEbs-1a, although capable of generating base editing in all targets with a five-member crRNA array containing two ncPAM crRNAs, exhibited reduced editing activity compared to duBE-1a. Furthermore, in Target 3 with a ncPAM GCTC, duBE-1a also possessed a certain level of activity ( Supplementary Figure S20C ), which is consistent with the previous study ( 72 ). Future work will involve searching for broad-spectrum mutations that better match duBE. Simultaneously, it is crucial to consider the trade-off between off-target effects and PAM preference, as stringent PAM recognition is known to reduce genome-wide off-target effects ( 73 ).
In conclusion, we revealed that the specific combination mode of cytosine deaminase, adenine deaminase, and dCas12a would guarantee the activities of all three proteins simultaneously, which enables multiplexed targeting and dual base editing. Furthermore, we found that interference in the crRNA array was a key factor that affects the performance of MultiduBE, and that introducing the synthetic separator sequence into the crRNA array significantly improved the multiplexed editing efficiency in both E . coli and B . subtilis . Finally, we demonstrated the value of MultiduBE for multiplexed in-situ mutagenesis, taking cell physiology reprogramming and metabolic regulation as examples. Given that mutations in the genome can be rapidly tracked using the crRNA array, MultiduBE will also be useful for genome-scale mutagenesis and multi-gene interaction analysis.
Reads from NGS of high-throughput targeted amplicon sequencing, whole genome resequencing, RNA-seq, and miRNA sequencing are submitted at Sequence Read Archive (PRJNA967093). The sequences of plasmid and crRNA array with the necessary annotations used in this study are uploaded to Benchling, and the according URLs are shown in Supplementary Note S2 and Supplementary Note S4 . Raw gel images, unedited micrographs, and source data generated by the BEAT software are deposited in Zenodo (10.5281/zenodo.10631289). Flow cytometry data is deposited at FlowRepository (FR-FCM-Z6AW).
Supplementary Data are available at NAR Online.
National Key R&D Program of China [2020YFA0908300, 2018YFA0900300]; National Natural Science Foundation of China [32021005, 32070085, 31930085, 32300064]; China National Postdoctoral Program for Innovative Talents [BX2021113]; Natural Science Foundation of Jiangsu Province [BK20221083]; Fundamental Research Funds for the Central Universities [USRP52019A, JUSRP221013, JUSRP124034, and JUSRP121010]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement . None declared.
Korry B.J. , Lee S.Y.E. , Chakrabarti A.K. , Choi A.H. , Ganser C. , Machan J.T. , Belenky P. Genotoxic agents produce stressor-specific spectra of spectinomycin resistance mutations based on mechanism of action and selection in Bacillus subtilis . Antimicrob. Agents Chemother. 2021 ; 65 : e00891-21 .
Google Scholar
Halperin S.O. , Tou C.J. , Wong E.B. , Modavi C. , Schaffer D.V. , Dueber J.E. CRISPR-guided DNA polymerases enable diversification of all nucleotides in a tunable window . Nature . 2018 ; 560 : 248 – 252 .
Esvelt K.M. , Carlson J.C. , Liu D.R. A system for the continuous directed evolution of biomolecules . Nature . 2011 ; 472 : 499 – 503 .
Ravikumar A. , Arzumanyan G.A. , Obadi M.K.A. , Javanpour A.A. , Liu C.C. Scalable, continuous evolution of genes at mutation rates above genomic error thresholds . Cell . 2018 ; 175 : 1946 – 1957 .
Tian R. , Rehm F.B.H. , Czernecki D. , Gu Y. , Zürcher J.F. , Liu K.C. , Chin J.W. Establishing a synthetic orthogonal replication system enables accelerated evolution in E. coli . Science . 2024 ; 383 : 421 – 426 .
Moore C.L. , Papa L.J.I. , Shoulders M.D. A processive protein chimera introduces mutations across defined DNA regions in vivo . J. Am. Chem. Soc. 2018 ; 140 : 11560 – 11564 .
Molina R.S. , Rix G. , Mengiste A.A. , Álvarez B. , Seo D. , Chen H. , Hurtado J.E. , Zhang Q. , García-García J.D. , Heins Z.J. et al. . In vivo hypermutation and continuous evolution . Nat. Rev. Methods Primer . 2022 ; 2 : 37 .
Wu Y. , Liu Y. , Lv X. , Li J. , Du G. , Liu L. Applications of CRISPR in a microbial cell factory: from genome reconstruction to metabolic network reprogramming . ACS Synth. Biol. 2020 ; 9 : 2228 – 2238 .
Anzalone A.V. , Koblan L.W. , Liu D.R. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors . Nat. Biotechnol. 2020 ; 38 : 824 – 844 .
Grünewald J. , Zhou R. , Lareau C.A. , Garcia S.P. , Iyer S. , Miller B.R. , Langner L.M. , Hsu J.Y. , Aryee M.J. , Joung J.K. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing . Nat. Biotechnol. 2020 ; 38 : 861 – 864 .
Sakata R.C. , Ishiguro S. , Mori H. , Tanaka M. , Tatsuno K. , Ueda H. , Yamamoto S. , Seki M. , Masuyama N. , Nishida K. et al. . Base editors for simultaneous introduction of C-to-T and A-to-G mutations . Nat. Biotechnol. 2020 ; 38 : 865 – 869 .
Zhang X. , Zhu B. , Chen L. , Xie L. , Yu W. , Wang Y. , Li L. , Yin S. , Yang L. , Hu H. et al. . Dual base editor catalyzes both cytosine and adenine base conversions in human cells . Nat. Biotechnol. 2020 ; 38 : 856 – 860 .
Li C. , Zhang R. , Meng X. , Chen S. , Zong Y. , Lu C. , Qiu J.-L. , Chen Y.-H. , Li J. , Gao C. 2020) Targeted, random mutagenesis of plant genes with dual cytosine and adenine base editors . Nat. Biotechnol. 38 : 875 – 882 .
Xie J. , Huang X. , Wang X. , Gou S. , Liang Y. , Chen F. , Li N. , Ouyang Z. , Zhang Q. , Ge W. et al. . ACBE, a new base editor for simultaneous C-to-T and A-to-G substitutions in mammalian systems . BMC Biol. 2020 ; 18 : 131 .
Shelake R.M. , Pramanik D. , Kim J.-Y. Improved dual base editor systems (iACBEs) for simultaneous conversion of adenine and cytosine in the bacterium Escherichia coli . mBio . 2023 ; 14 : e02296-22 .
Yuan Q. , Gao X. Multiplex base- and prime-editing with drive-and-process CRISPR arrays . Nat. Commun. 2022 ; 13 : 2771 .
McCarty N.S. , Graham A.E. , Studená L. , Ledesma-Amaro R. Multiplexed CRISPR technologies for gene editing and transcriptional regulation . Nat. Commun. 2020 ; 11 : 1281 .
Wu Y. , Li Y. , Jin K. , Zhang L. , Li J. , Liu Y. , Du G. , Lv X. , Chen J. , Ledesma-Amaro R. et al. . CRISPR–dCas12a-mediated genetic circuit cascades for multiplexed pathway optimization . Nat. Chem. Biol. 2023 ; 19 : 367 – 377 .
Shaw W.M. , Studená L. , Roy K. , Hapeta P. , McCarty N.S. , Graham A.E. , Ellis T. , Ledesma-Amaro R. Inducible expression of large gRNA arrays for multiplexed CRISPRai applications . Nat. Commun. 2022 ; 13 : 4984 .
Li X. , Wang Y. , Liu Y. , Yang B. , Wang X. , Wei J. , Lu Z. , Zhang Y. , Wu J. , Huang X. et al. . Base editing with a Cpf1-cytidine deaminase fusion . Nat. Biotechnol. 2018 ; 36 : 324 – 327 .
Wang X. , Ding C. , Yu W. , Wang Y. , He S. , Yang B. , Xiong Y.-C. , Wei J. , Li J. , Liang J. et al. . Cas12a base editors induce efficient and specific editing with low DNA damage response . Cell Rep. 2020 ; 31 : 107723 .
Kempton H.R. , Love K.S. , Guo L.Y. , Qi L.S. Scalable biological signal recording in mammalian cells using Cas12a base editors . Nat. Chem. Biol. 2022 ; 18 : 742 – 750 .
Chen F. , Lian M. , Ma B. , Gou S. , Luo X. , Yang K. , Shi H. , Xie J. , Ge W. , Ouyang Z. et al. . Multiplexed base editing through Cas12a variant-mediated cytosine and adenine base editors . Commun. Biol. 2022 ; 5 : 1163 .
Gaillochet C. , Peña Fernández A. , Goossens V. , D’Halluin K. , Drozdzecki A. , Shafie M. , Van Duyse J. , Van Isterdael G. , Gonzalez C. , Vermeersch M. et al. . Systematic optimization of Cas12a base editors in wheat and maize using the ITER platform . Genome Biol. 2023 ; 24 : 6 .
Cheng Y. , Zhang Y. , Li G. , Fang H. , Sretenovic S. , Fan A. , Li J. , Xu J. , Que Q. , Qi Y. CRISPR-Cas12a base editors confer efficient multiplexed genome editing in rice . Plant Commun. 2023 ; 4 : 100601 .
Wu Y. , Chen T. , Liu Y. , Tian R. , Lv X. , Li J. , Du G. , Chen J. , Ledesma-Amaro R. , Liu L. Design of a programmable biosensor-CRISPRi genetic circuits for dynamic and autonomous dual-control of metabolic flux in Bacillus subtilis . Nucleic Acids Res. 2020 ; 48 : 996 – 1009 .
Li Y. , Wu Y. , Liu Y. , Li J. , Du G. , Lv X. , Liu L. A genetic toolkit for efficient production of secretory protein in Bacillus subtilis . Bioresour. Technol. 2022 ; 363 : 127885 .
Wu Y. , Liu Y. , Lv X. , Li J. , Du G. , Liu L. CAMERS-B: cRISPR/Cpf1 assisted multiple-genes editing and regulation system for Bacillus subtilis . Biotechnol. Bioeng. 2020 ; 117 : 1817 – 1825 .
Xu L. , Liu Y. , Han R. BEAT: a python program to quantify base editing from sanger sequencing . CRISPR J. 2019 ; 2 : 223 – 229 .
Bae S. , Park J. , Kim J.-S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases . Bioinformatics . 2014 ; 30 : 1473 – 1475 .
Yang H. , Yu H. , Shen Z. A novel high-throughput and quantitative method based on visible color shifts for screening Bacillus subtilis THY-15 for surfactin production . J. Ind. Microbiol. Biotechnol. 2015 ; 42 : 1139 – 1147 .
Sun G. , Wu Y. , Huang Z. , Liu Y. , Li J. , Du G. , Lv X. , Liu L. Directed evolution of diacetylchitobiose deacetylase via high-throughput droplet sorting with a novel, bacteria-based biosensor . Biosens. Bioelectron. 2023 ; 219 : 114818 .
You J. , Yang C. , Pan X. , Hu M. , Du Y. , Osire T. , Yang T. , Rao Z. Metabolic engineering of Bacillus subtilis for enhancing riboflavin production by alleviating dissolved oxygen limitation . Bioresour. Technol. 2021 ; 333 : 125228 .
Mack M. , van Loon A.P.G.M. , Hohmann H.-P. Regulation of riboflavin biosynthesis in Bacillus subtilis is affected by the activity of the flavokinase/flavin adenine dinucleotide synthetase encoded by ribC . J. Bacteriol. 1998 ; 180 : 950 – 955 .
Liu X. , Liu X. , Zhou C. , Lv J. , He X. , Liu Y. , Xie H. , Wang B. , Lv X. , Tang L. et al. . Engineered FnCas12a with enhanced activity through directional evolution in human cells . J. Biol. Chem. 2021 ; 296 : 100394 .
Meliawati M. , Schilling C. , Schmid J. Recent advances of Cas12a applications in bacteria . Appl. Microbiol. Biotechnol. 2021 ; 105 : 2981 – 2990 .
Bandyopadhyay A. , Kancharla N. , Javalkote V.S. , Dasgupta S. , Brutnell T.P. CRISPR-Cas12a (Cpf1): a versatile tool in the plant genome editing tool box for agricultural advancement . Front. Plant Sci. 2020 ; 11 : 584151 .
Li A. , Mitsunobu H. , Yoshioka S. , Suzuki T. , Kondo A. , Nishida K. Cytosine base editing systems with minimized off-target effect and molecular size . Nat. Commun. 2022 ; 13 : 4531 .
Cheng T.-L. , Li S. , Yuan B. , Wang X. , Zhou W. , Qiu Z. Expanding C–T base editing toolkit with diversified cytidine deaminases . Nat. Commun. 2019 ; 10 : 3612 .
Yan D. , Ren B. , Liu L. , Yan F. , Li S. , Wang G. , Sun W. , Zhou X. , Zhou H. High-efficiency and multiplex adenine base editing in plants using new TadA variants . Mol. Plant . 2021 ; 14 : 722 – 731 .
Neugebauer M.E. , Hsu A. , Arbab M. , Krasnow N.A. , McElroy A.N. , Pandey S. , Doman J.L. , Huang T.P. , Raguram A. , Banskota S. et al. . Evolution of an adenine base editor into a small, efficient cytosine base editor with low off-target activity . Nat. Biotechnol. 2023 ; 41 : 673 – 685 .
Lam D.K. , Feliciano P.R. , Arif A. , Bohnuud T. , Fernandez T.P. , Gehrke J.M. , Grayson P. , Lee K.D. , Ortega M.A. , Sawyer C. et al. . Improved cytosine base editors generated from TadA variants . Nat. Biotechnol. 2023 ; 41 : 686 – 697 .
Zetsche B. , Heidenreich M. , Mohanraju P. , Fedorova I. , Kneppers J. , Degennaro E.M. , Winblad N. , Choudhury S.R. , Abudayyeh O.O. , Gootenberg J.S. et al. . Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array . Nat. Biotechnol. 2017 ; 35 : 31 – 34 .
Magnusson J.P. , Rios A.R. , Wu L. , Qi L.S. Enhanced Cas12a multi-gene regulation using a CRISPR array separator . eLife . 2021 ; 10 : e66406 .
Grünewald J. , Zhou R. , Garcia S.P. , Iyer S. , Lareau C.A. , Aryee M.J. , Joung J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors . Nature . 2019 ; 569 : 433 – 437 .
Galinier A. , Foulquier E. , Pompeo F. Metabolic control of cell elongation and cell division in Bacillus subtilis . Front. Microbiol. 2021 ; 12 : 697930 .
Blair J.M.A. , Webber M.A. , Baylay A.J. , Ogbolu D.O. , Piddock L.J.V. Molecular mechanisms of antibiotic resistance . Nat. Rev. Microbiol. 2015 ; 13 : 42 – 51 .
Maughan H. , Galeano B. , Nicholson W.L. Novel rpoB mutations conferring rifampin resistance on Bacillus subtilis : global effects on growth, competence, sporulation, and germination . J. Bacteriol. 2004 ; 186 : 2481 – 2486 .
Tojo S. , Kim J.-Y. , Tanaka Y. , Inaoka T. , Hiraga Y. , Ochi K. The mthA mutation conferring low-level resistance to streptomycin enhances antibiotic production in Bacillus subtilis by increasing the S-adenosylmethionine pool size . J. Bacteriol. 2014 ; 196 : 1514 – 1524 .
Bechhofer D.H. , Stasinopoulos S.J. tetA (L) mutants of a tetracycline-sensitive strain of Bacillus subtilis with the polynucleotide phosphorylase gene deleted . J. Bacteriol. 1998 ; 180 : 3470 – 3473 .
Ilina E. , Malakhova M. , Bodoev I. , Filimonova A. , Oparina N. , Govorun V. Mutation in ribosomal protein S5 leads to spectinomycin resistance in Neisseria gonorrhoeae . Front. Microbiol. 2013 ; 4 : 51849 .
Chen S.-C. , Hu L.-H. , Zhu X.-Y. , Yin Y.-P. Gonococcal urethritis caused by a multidrug resistant Neisseria gonorrhoeae strain with high-level resistance to spectinomycin in China . Emerg. Microbes Infect. 2020 ; 9 : 517 – 519 .
He X. , Miao V. , Baltz R.H. Spectinomycin resistance in rpsE mutants is recessive in Streptomyces roseosporus . J. Antibiot. 2005 ; 58 : 284 – 288 .
Criswell D. , Tobiason V.L. , Lodmell J.S. , Samuels D.S. Mutations conferring aminoglycoside and spectinomycin resistance in Borrelia burgdorferi . Antimicrob. Agents Chemother. 2006 ; 50 : 445 – 452 .
Sun Y.-J. , Luo J.-T. , Wong S.-Y. , Lee A.S.G. Analysis of rpsL and rrs mutations in Beijing and non-Beijing streptomycin-resistant mycobacterium tuberculosis isolates from Singapore . Clin. Microbiol. Infect. 2010 ; 16 : 287 – 289 .
Hosaka T. , Xu J. , Ochi K. Increased expression of ribosome recycling factor is responsible for the enhanced protein synthesis during the late growth phase in an antibiotic-overproducing Streptomyces coelicolor ribosomal rpsL mutant . Mol. Microbiol. 2006 ; 61 : 883 – 897 .
Andersson D.I. , Hughes d. Antibiotic resistance and its cost: is it possible to reverse resistance? . Nat. Rev. Microbiol. 2010 ; 8 : 260 – 271 .
Xia L. , Wen J. Available strategies for improving the biosynthesis of surfactin: a review . Crit. Rev. Biotechnol. 2022 ; 43 : 1111 – 1128 .
Wu Q. , Zhi Y. , Xu Y. Systematically engineering the biosynthesis of a green biosurfactant surfactin by Bacillus subtilis 168 . Metab. Eng. 2019 ; 52 : 87 – 97 .
Wang M. , Yu H. , Li X. , Shen Z. Single-gene regulated non-spore-forming Bacillus subtilis : construction, transcriptome responses, and applications for producing enzymes and surfactin . Metab. Eng. 2020 ; 62 : 235 – 248 .
You J. , Pan X. , Yang C. , Du Y. , Osire T. , Yang T. , Zhang X. , Xu M. , Xu G. , Rao Z. Microbial production of riboflavin: biotechnological advances and perspectives . Metab. Eng. 2021 ; 68 : 46 – 58 .
Hao W. , Cui W. , Cheng Z. , Han L. , Suo F. , Liu Z. , Zhou L. , Zhou Z. Development of a base editor for protein evolution via in situ mutation in vivo . Nucleic Acids Res. 2021 ; 49 : 9594 – 9605 .
Hao W. , Cui W. , Suo F. , Han L. , Cheng Z. , Zhou Z. 2022) Construction and application of an efficient dual-base editing platform for Bacillus subtilis evolution employing programmable base conversion . Chem. Sci. 13 : 14395 – 14409 .
Komor A.C. , Kim Y.B. , Packer M.S. , Zuris J.A. , Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage . Nature . 2016 ; 533 : 420 – 424 .
Tong H. , Wang X. , Liu Y. , Liu N. , Li Y. , Luo J. , Ma Q. , Wu D. , Li J. , Xu C. et al. . Programmable A-to-Y base editing by fusing an adenine base editor with an N-methylpurine DNA glycosylase . Nat. Biotechnol. 2023 ; 41 : 1080 – 1084 .
Zhao D. , Li J. , Li S. , Xin X. , Hu M. , Price M.A. , Rosser S.J. , Bi C. , Zhang X. Glycosylase base editors enable C-to-A and C-to-G base changes . Nat. Biotechnol. 2021 ; 39 : 35 – 40 .
Kurt I.C. , Zhou R. , Iyer S. , Garcia S.P. , Miller B.R. , Langner L.M. , Grünewald J. , Joung J.K. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells . Nat. Biotechnol. 2021 ; 39 : 41 – 46 .
Yamano T. , Nishimasu H. , Zetsche B. , Hirano H. , Slaymaker I.M. , Li Y. , Fedorova I. , Nakane T. , Makarova K.S. , Koonin E.V. et al. . Crystal structure of Cpf1 in complex with guide RNA and target DNA . Cell . 2016 ; 165 : 949 – 962 .
Aman R. , Syed M.M. , Saleh A. , Melliti F. , Gundra S.R. , Wang Q. , Marsic T. , Mahas A. , Mahfouz M.M. Peptide nucleic acid-assisted generation of targeted double-stranded DNA breaks with T7 endonuclease I . Nucleic Acids Res. 2024 ; gkae148 .
Yu W. , Xu X. , Jin K. , Liu Y. , Li J. , Du G. , Lv X. , Liu L. Genetically encoded biosensors for microbial synthetic biology: from conceptual frameworks to practical applications . Biotechnol. Adv. 2023 ; 62 : 108077 .
Ma S. , Liao K. , Li M. , Wang X. , Lv J. , Zhang X. , Huang H. , Li L. , Huang T. , Guo X. et al. . Phase-separated DropCRISPRa platform for efficient gene activation in mammalian cells and mice . Nucleic Acids Res. 2023 ; 51 : 5271 – 5284 .
Chen Z. , Sun J. , Guan Y. , Li M. , Lou C. , Wu B. Engineered DNase-inactive Cpf1 variants to improve targeting scope for base editing in E. coli . Synth. Syst. Biotechnol. 2021 ; 6 : 326 – 334 .
Zhou J. , Chen P. , Wang H. , Liu H. , Li Y. , Zhang Y. , Wu Y. , Paek C. , Sun Z. , Lei J. et al. . Cas12a variants designed for lower genome-wide off-target effect through stringent PAM recognition . Mol. Ther. 2022 ; 30 : 244 – 255 .
Email alerts
Citing articles via.
- Editorial Board
Affiliations
- Online ISSN 1362-4962
- Print ISSN 0305-1048
- Copyright © 2024 Oxford University Press
- About Oxford Academic
- Publish journals with us
- University press partners
- What we publish
- New features
- Open access
- Institutional account management
- Rights and permissions
- Get help with access
- Accessibility
- Advertising
- Media enquiries
- Oxford University Press
- Oxford Languages
- University of Oxford
Oxford University Press is a department of the University of Oxford. It furthers the University's objective of excellence in research, scholarship, and education by publishing worldwide
- Copyright © 2024 Oxford University Press
- Cookie settings
- Cookie policy
- Privacy policy
- Legal notice
This Feature Is Available To Subscribers Only
Sign In or Create an Account
This PDF is available to Subscribers Only
For full access to this pdf, sign in to an existing account, or purchase an annual subscription.
An NIH genetics study targets a long-standing challenge: Diversity
Good morning. I’m Lauren Sausser, a KFF Health News reporter based in Charleston, S.C., where my allergy shots seemed to stop working around mid-February. I cover health-care news across the South, often issues related to health equity. Send story ideas to [email protected] .
Not a subscriber? Sign up here.
Today’s edition: Medicare is headed for insolvency, though the economy has bought it some extra time, according to a new report. The Biden administration says a public education campaign on vaccination saved tens of thousands of lives. But first …
An NIH program could be a model for more diverse clinical studies
In his 2015 State of the Union address, President Barack Obama announced a precision medicine initiative that would later be known as the All of Us program. The research, now well underway at the National Institutes of Health , aims to analyze the DNA of at least 1 million people across the United States to build a diverse health database.
The key word there is “diverse.” So far, the program has collected more than 560,000 DNA samples, and nearly half of participants identify as being part of a racial or ethnic minority group.
NIH researchers strategically partnered with community health centers, faith-based groups, and Black fraternities and sororities to recruit people who have been historically underrepresented in biomedical research.
“We are actually looking to overrepresent” these previously marginalized groups, explained Martin Mendoza , director of health equity for All of Us, which will continue to enroll participants through at least 2026, when researchers intend to evaluate the next phases of the project.
Their success to date is remarkable for a few reasons. Participation in biomedical research is typically low in diversity. And when it comes to genetics research specifically, diversity has been nearly nonexistent.
Since the completion more than 20 years ago of the Human Genome Project , which mapped most human genes for the first time, nearly 90 percent of genomics studies have been conducted using DNA from participants of European descent, research shows .
Humans of all races and ethnicities are 99 percent genetically identical. But even small differences in our DNA can have a profound impact on our health.
Here’s an example: A few years ago, researchers found that some Black patients had been misdiagnosed with a potentially fatal heart condition called hypertrophic cardiomyopathy because they’d tested positive for genetic variants that were thought to be harmful. But it turns out the variants, more common among Black Americans than among White Americans , are likely harmless. The diagnosis, though, is life-altering — patients with hypertrophic cardiomyopathy have traditionally been discouraged from competing in sports, for example.
Such misdiagnoses can be avoided if “even modest numbers of people from diverse populations are included in sequence databases,” NIH wrote.
Easier said than done. A genetics research project underway in South Carolina called In Our DNA SC is struggling to recruit enough Black participants . The scientists behind the project said two years ago they aimed to collect samples reflecting the diversity of the state, where 27 percent of residents identify as Black or African American.
“We’d like to be a lot more diverse,” said Daniel Judge , principal investigator for the study and a cardiovascular genetics specialist at the Medical University of South Carolina .
To date, only about 12 percent of participants who provided socio-demographic data identify as Black. An additional 5 percent identify as belonging to another racial minority.
KFF Health News is a national newsroom that produces in-depth journalism about health issues and is one of the core operating programs at KFF — an independent source of health policy research, polling, and journalism.
Agency alert
Medicare and social security finances look grim, trustees say.
Medicare and Social Security will run out of money in just over a decade if Congress doesn’t act, a new report warned Monday, according to The Post’s Jacob Bogage and Julie Zauzmer Weil . The trustees for the massive retirement programs project that Social Security will be insolvent by 2035, and Medicare by 2036, which would force benefit cuts – unless Congress acts.
The good-ish news : That’s better than many experts had expected — last year, federal actuaries said the programs could go belly-up sooner.
In a separate FAQ , Julie and Scott Sowers reported that the trustees upgraded their outlook for the trust fund that pays for Medicare hospital benefits, projecting that it has enough money to cover full benefits until 2036, five years longer than forecast last year.
Growth has been robust and employment rates have been high, meaning more people are paying taxes into the trust fund. The Medicare hospital fund also spent less in 2023 than the trustees expected.
The bad news : After 2036, without policy changes, hospital coverage would be cut by 11 percent.
Lawmakers, with their eyes on November’s elections, say they could face a rare window to enact sweeping fiscal reforms in 2025. The next year will bring major fiscal policy decisions for whoever wins the election and for Congress. Read Jacob and Julie’s story here . Read Julie and Scott’s FAQ here .
From our notebook
Hhs-backed study: 'we can do this' vaccine campaign saved lives, money.
The Post’s Dan Diamond has this update:
The Biden administration’s public education campaign for coronavirus vaccines “saved more than 50,000 lives and prevented hundreds of thousands of hospitalizations and millions of COVID-19 cases, representing hundreds of billions of dollars in benefits in less than one year,” Department of Health and Human Services staff and Fors Marsh consultants conclude in the American Journal of Preventive Medicine .
The study reviewed HHS efforts between April 2021 and March 2022, concluding that the vaccine campaign encouraged about 22 million people to get vaccinated across the year, preventing about 244,000 hospitalizations, including during the delta and omicron variant spikes. Fors Marsh , a research consultancy group, worked with HHS on the campaign, which included advertising on digital, TV, radio, print and other platforms.
Taking a long-term view: Publishing the findings helps demonstrate the value of public awareness campaigns, said Joshua Peck , a former Biden administration official who co-authored the study.
He noted that the Obama administration's findings on the benefits of Affordable Care Act outreach and advertising were denied by the Trump administration, which moved to cut spending on ACA outreach and ads .
“By putting this into the public domain, we are doing our very best to make sure that the work of other people who do public education work can't be denied by others,” Peck said in an interview.
HHS Secretary Xavier Becerra touted the findings , calling the campaign an “indispensable part” of the Biden administration’s pandemic response. “We will no doubt use what we learned in this campaign to further improve our public health efforts in the future,” he said in a statement.
Industry Rx
Fda-approved sickle cell therapies move forward.
The Post’s Carolyn Y. Johnson filed this report:
It’s a historic moment for sickle cell disease , a genetic condition that affects more than 100,000 people in the United States, causing debilitating pain episodes and cutting decades off people’s lives.
Federal regulators last year gave the green light to two sickle cell gene therapies , and this week, both companies announced that they have begun the first steps of multistep procedures that will unfold over months.
One of the companies, Vertex Pharmaceuticals , reported that stem cells have been collected from a handful of patients. The other, Bluebird Bio , announced Monday that doctors at Children’s National Hospital in D.C. harvested stem cells from a 12-year-old patient .
Those stem cells will be genetically modified at specialized laboratories and tweaked so they produce a form of hemoglobin, the oxygen-carrying protein in red blood cells, that doesn’t collapse into the rigid, sickle-shaped cells that are the hallmark of the disease. Patients then receive chemotherapy, before receiving their modified stem cells.
Both therapies had profound effects for patients in clinical trials, but they are pricey. Bluebird Bio’s therapy carries a list price of $3.1 million and Vertex’s is $2.2 million — raising questions about how to give patients equitable access to the therapy. About half of patients with sickle cell are covered by Medicaid , and the Biden administration announced in January that sickle cell disease would be the first focus of a new model aimed at expanding access to gene therapy.
On the Hill
Sen. Bernie Sanders (I-Vt.) announced Monday that he will run for reelection this year, ending months of speculation, The Post’s Amy B Wang and Liz Goodwin report . Sanders, 82, will be seeking a fourth term.
As chairman of the Committee on Health, Education, Labor and Pensions , he has cut an energetic figure — shutting down an almost-physical altercation between a union leader and a senator with a wagging finger and grilling pharmaceutical CEOs last year.
In his announcement, Sanders flagged his work on drug costs, among other achievements. He also said more work is needed to “end the absurdity of Americans paying, by far, the highest prices in the world for prescription drugs.”
Meanwhile on the other side of the Capitol, Rep. Jennifer Wexton (D-Va.) used assistive technology to speak on the floor of the House yesterday, The Post’s Mariana Alfaro reports .
Wexton was diagnosed with progressive supranuclear palsy (PSP) last year, which has largely affected her ability to speak, hear and move. Wexton a nnounced in September that she would not run for reelection; she now uses a text-to-voice application. PSP, a rare neurological condition, typically progresses rapidly and has no cure.
Health reads
A gene long thought to just raise the risk for Alzheimer's may cause some cases (By Lauren Neergaard | AP)
Could better asthma inhalers help patients, and the planet too? (By Martha Bebinger | NPR)
Facing Unchecked Syphilis Outbreak, Great Plains Tribes Sought Federal Help. Months Later, No One Has Responded. (By Anna Maria Barry-Jester)
Thanks for reading. See you tomorrow!


College of Biological Sciences
One cbs student’s mission to inspire and uplift the next generation of scientists.

- by Liana Wait
- May 09, 2024
Jessica Bolivar, a graduate student in the Biochemistry, Molecular, Cellular and Developmental Biology (BMCDB) Graduate Group, knows firsthand the difference that one person’s mentorship can make.
During her time at UC Davis, Bolivar has made it her mission to give back and inspire the next generation of scientists by balancing her research with a slew of community-uplifting and diversity, equity and inclusion initiatives.
“One person changed my whole career path,” Bolivar said. “That’s where my passion comes from with these initiatives—I just want to inspire the next generation of students.”

Her parents’ legacy
Bolivar grew up in San Pablo, California in a large but close-knit family that taught her to prioritize community and education. She and her siblings were the first in their family to graduate college, and Bolivar says she was inspired to strive academically by her parents, particularly her father.
“My dad unfortunately wasn’t able to witness us graduate because he passed away, but he’s the reason I’m here—I do everything because my parents made sacrifices in order for me to go to school,” said Bolivar.
A winding path to research
“I got into science by accident,” Bolivar said.

Bolivar always wanted to help people. As an undergraduate at San Francisco State University (SFSU), she aspired to become a medical doctor, but when her father got sick, she wasn’t able to maintain her grades. After graduating, she worked for several years in the Housing Department at her alma mater while taking courses to boost her GPA—all while l dreaming of med school. Then, a chance conversation convinced her to give research a try.
“It was my former cell biology professor who got me back into school,” Bolivar said. “He told me, you can help people in other ways—you can help people in research.”
Bolivar went on to complete a master’s degree in cell and molecular biology at SFSU before moving to Davis, where she is currently pursuing her Ph.D. “I just fell in love with research,” she said. “’It’s a journey where you’re exploring new territory.”
Cell imaging—a window into the cell’s stress response
In the BMCDB graduate group, Bolivar is investigating the molecular mechanisms that cells use to deal with stress in the lab of Christopher Fraser , a professor in the Department of Molecular and Cellular Biology. In the predominately biochemistry-based lab, Bolivar is taking a different angle to examine cell stress—by drawing on her passion and experience with cell imaging.
“I love cell biology, I love imaging,” Bolivar said. “I just feel at peace when I can see something. It’s so fascinating to watch a process and then disrupt it and see what happens.”
Fraser, Bolivar’s advisor, says that her dedication to science and fostering an inclusive workplace culture have left a deep impression.
“Within the laboratory, her creation of an innovative assay for real-time monitoring of the cellular stress response has revolutionized our comprehension of this intricate process,” Fraser said. “Beyond the laboratory, Jessica's altruistic efforts to champion underrepresented scientists at UC Davis serve as a beacon of inspiration.”
Giving back—DEIJ work at Davis
With the support and mentorship of Ben Montpetit , the Chair of the Biochemistry, Molecular, Cell and Developmental Biology graduate group, Bolivar has used her time at Davis to give back in various ways.
“Jessica is a wonderful person who simply cares about everyone,” Montpetit said. “She has committed countless hours to building community here at UC Davis. Her efforts have created changes within BMCDB that impact how we operate, which will continue long after Jessica graduates and moves on from UC Davis.”
As the co-chair of outreach for the UC Davis Diversity, Equity, Inclusion Committee from 2020 to 2022, Bolivar traveled to California State Universities where she coordinated and spoke at colloquiums and workshops for undergraduate and master’s students.

Along with graduate students, Jasmine Esparza and Cuauhtemoc Gonzalez, Bolivar co-founded theirSTORY in 2023, a seminar series that celebrates underrepresented scientists. The inspirational talks focus on the narratives and journeys of UC Davis faculty and staff with diverse identities and backgrounds.
“It’s important to hear from people who have already succeeded in these positions, and to learn about their journeys and identities,” said Bolivar. “I’m first generation, so I didn’t have mentors or family members that have already been in these positions.”
Bolivar also co-founded the California Emerging Scientist Workshop, a 5-day workshop that UC Davis premiered in August 2023 in partnership with the Advanced Imaging Center at HHMI Janelia.
The workshop, which brought together 24 undergraduate and master’s students from diverse backgrounds and introduced them to the fundamentals of microscopy and analysis, was inspired by Bolivar’s own experience at a two-week microscopy workshop that she had attended at Janelia. “I just thought, wow, I wish I had this information when I was a younger career scientist,” Bolivar said.
Participants described the experience as a “gamechanger” that made them more comfortable as scientists, Bolivar said. “It was a lot of positivity.”

Aspiring to inspire
Bolivar, who is set to graduate this spring, is still exploring her immediate next steps but plans to continue promoting diversity and equity in science. Ultimately, she dreams of setting up a science camp for kids with disabilities.
“I have a disability, and I think it’s really important that we promote equity,” said Bolivar. “Anywhere I can impact and inspire the next generation, that’s where I want to be.”
Media Resources
- Liana Wait is a freelance science writer based in Philadelphia. She has a Ph.D. in ecology and evolutionary biology and specializes in writing about the life sciences.
Primary Category
Secondary categories.
IMAGES
VIDEO
COMMENTS
Romiguier, J. et al. Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature 515, 261-263 (2014). This study shows a comparative analysis of patterns of ...
Genetic diversity and adaptive potential within populations of all [wild and domestic] species is safeguarded, and all genetically distinct populations are maintained by 2030, and at least 99% of genetic diversity within populations is maintained by 2050 ... Taft HR, McCoskey DN, Miller JM, et al. Research-management partnerships: an ...
Genetic diversity within and between plant species allows plant breeders to select superior genotypes, which can then be used for the development of genetic stock for hybridization programs or the release of a crop variety . ... Digitized molecular data are vital to numerous aspects of scientific research and genetic resource use. Substantial ...
Why do genomic research in diverse populations? Motivations to conduct research in the context of genetic diversity are numerous. Increased inclusion facilitates the understanding of health disparities, new discoveries in biology, more accurate matching of diverse patients with safe and effective treatments, improved interpretation of genetic tests, and better tracing of human history.
Research Article. Share on. Insights into human genetic variation and population history from 929 diverse genomes. ... To add to our understanding of human genetic diversity, Bergström et al. generated whole-genome sequences surveying individuals in the Human Genome Diversity Project, which is a panel of global populations that has been ...
The aim of this study is to: (i) re-examine the goals of the management of genetic diversity in breeding schemes, and the molecular genetic parameters that may be incorporated into these goals; and (ii) compare alternative genomic- and pedigree-based measures of inbreeding and relationships for addressing the goals.
Genetic diversity among and within populations of all species is necessary for people and nature to survive and thrive in a changing world. Over the past three years, commitments for conserving genetic diversity have become more ambitious and specific under the Convention on Biological Diversity's (CBD) draft post-2020 global biodiversity framework (GBF). This Perspective article comments on ...
Genomic research has evolved from seeking to understand the fundamentals of the human genetic code to examining the ways in which this code varies among people, and then applying this knowledge to ...
And when it comes to genetics research specifically, diversity has been nearly nonexistent. Since the completion more than 20 years ago of the Human Genome Project, which mapped most human genes for the first time, nearly 90 percent of genomics studies have been conducted using DNA from participants of European descent, research shows.
Explore the latest full-text research PDFs, articles, conference papers, preprints and more on GENETIC DIVERSITY. Find methods information, sources, references or conduct a literature review on ...
Genetic diversity helps to adapt to environmental variability. Organisms live in complex environment that vary in spatial and temporal scale and. is characterized by several factors such as ...
Decreased population genetic diversity can be associated with declines in population fitness (e.g., [1, 2]).These declines are thought to involve components of the so called genetic 'extinction vortex', which directly ties losses in population genetic diversity to increased extinction risk [].These losses cause a decrease in individual fitness through the expression of inbreeding depression ...
Phenotypic characterization of sorghum seedlings and expression patterns of SNAC1 and DREB1A genes under water-stress: an insight towards developing resilient cultivars. Genetic Resources and Crop Evolution covers all aspects of plant genetic resources research with original articles in taxonomical, morphological, ...
A comprehensive strategy for the conservation of forest tree genetic diversity: an example with the protected Pinus nigra subsp. salzmannii (Dunal) Franco in France. C. Scotti-Saintagne; ... Research Article Open access 27 September 2023 Pages: 305 - 318 Conservation genomics of the threatened Trispot Darter (Etheostoma trisella) Kayla M. Fast ...
Genetic diversity is important because it gives species a better chance of survival. However, genetic diversity can be lost when populations get smaller and isolated, which decreases a species' ability to adapt and survive. In this article, we explore the importance of genetic diversity, discuss how it is formed and maintained in wild ...
Lack of diversity in genetics research has real health care implications. Since the completion more than 20 years ago of the Human Genome Project, which mapped most human genes for the first time, close to 90% of genomics studies have been conducted using DNA from participants of European descent, research shows. And while human beings of all ...
Abstract. Mutagenesis driving genetic diversity is vital for understanding and engineering biological systems. However, the lack of effective methods to generate in-situ mutagenesis in multiple genomic loci combinatorially limits the study of complex biological functions. Here, we design and construct MultiduBE, a dCas12a-based multiplexed dual-function base editor, in an all-in-one plasmid ...
This Special Issue on "Genetic Diversity, Population Structure, and Ancestral Origin of Livestock" aims to embrace any aspects of origin of livestock species regarding their links to wild relatives and highlight their shared ancestral genome components based on using molecular markers. ... Research articles, review articles as well as short ...
A genetics research project underway in South Carolina called In Our DNA SC is struggling to recruit enough Black participants. The scientists behind the project said two years ago they aimed to ...
Genetic diversity analysis of germplasm resources is a key component of identifying and evaluatinggermplasm resources and plays a crucial role in the discovery of excellent germplasms and important functional genes (Chen et al. 2007). Studies evaluating the genetic diversity of tea tree resources based on morphological traits and biochemical ...
Jessica Bolivar, a graduate student in the Biochemistry, Molecular, Cellular and Developmental Biology (BMCDB) Graduate Group, knows firsthand the difference that one person's mentorship can make. During her time at UC Davis, Bolivar has made it her mission to give back and inspire the next generation of scientists by balancing her research with a slew of community-uplifting and diversity ...